Download

1 / 32
320 likes | 480 Views
Colorimetric detection of cys using gold nanoparitlces aggregation 组会报告. 2012 年 11 月 9 日 李世滋. 1 手性相关文献 2 近期 GNPs 检测 cys 的工作进展. 具有磁核的纳米粒子在其表面同时有手性配体同时具有磁性和手性识别的能力 ,以此, 建立一种通用的手性磁性材料。

E N D
Colorimetric detection of cys using gold nanoparitlces aggregation 组会报告 2012年11月9日 李世滋 1手性相关文献 2近期GNPs检测cys的工作进展
具有磁核的纳米粒子在其表面同时有手性配体同时具有磁性和手性识别的能力,以此,建立一种通用的手性磁性材料。具有磁核的纳米粒子在其表面同时有手性配体同时具有磁性和手性识别的能力,以此,建立一种通用的手性磁性材料。 纤维素-2,3-二(3,5-二甲基苯基氨基甲酸酯)(CBDMPC),作为HPLC最有效的手性固定相,选择性固定在包硅磁球表面,形成手性磁微球Fe3O4@SiO2@CBDMPC(CMM)。之后,成功将CBDMPC固定在包硅微球的表面,用IR和CD谱表征。 最后,将磁性纳米材料直接手性分离领域中。分离结果证明CMM对不同消旋物聚有手性识别能力,证明了它作为手性分离的一种有潜力的材料的可行性。此外,外加磁场使得CMM的回收非常方便。
在无水吡啶中溶胀100℃,5h,活化羟基。 首先,纤维素之后加入过量的三苯甲基氯,使仅与6位的初级羟基反应形成三苯甲基醚。24h后,加入过量的3,5-二甲基苯异氰酸,形成在2-和3-位有羟基的氨基甲酸酯。该反应在100℃继续反应24h。 中间物,纤维素-2,3-二(3,5--二甲基苯基氨基甲酸酯)-6-O-三苯甲基因为在甲醇中不溶解而游离出来,在含有少量的盐酸的过量甲醇中再分散室温24h,除去三苯甲基氯,得到终产物CBDMPC。
1,6-己二异氰酸 为间隔臂:连接羟基和异氰酸→氨基甲酰基 70nm CBDMPC 将磁球在含有60mL苯的烧瓶中超声分散。 为了在磁球表面固化CBDMPC,将合成的CBDMPC溶解于20mL无水吡啶中,之后机械搅拌90℃加入上述MNPs分散液。搅拌1h后,加入100μL的1,6-己二异氰酸酯,反应在90℃进行12h。得到的溶液室温条件下冷却,通过磁性倾析手性手性磁性纳米粒子,用甲醇洗涤三次。最后产物70℃真空干燥12h。
消旋样品制备:在25mL乙醇中分别,溶解5种不同的消旋物。之后再以上5种消旋溶液中分别加入150mgMNPs。室温涡旋震荡5min,加磁场60s内得到上清液,用自动数字旋光计分析,其他消旋样品同上。消旋样品制备:在25mL乙醇中分别,溶解5种不同的消旋物。之后再以上5种消旋溶液中分别加入150mgMNPs。室温涡旋震荡5min,加磁场60s内得到上清液,用自动数字旋光计分析,其他消旋样品同上。 为了研究分离过程,择5种消旋的安息香甲酯的一种,分离结果用HPLC分析。简单说,通过在乙醇中稀释得到浓度为10μg/ml的消旋样品。用HPLC分析,在试管中加入1mL的消旋样品(10μg/ml),随后加入100mg合成的手性磁性微球。混合液在室温条件下涡旋震荡5min。之后在磁场的帮助下,溶液与CMM在60s内得到分离,收集上清液进行HPLC分析。之后剩余溶液与CMM保持多余15min,上清液用HPLC分析。 回收: 用CMM分离5种消旋物后(总用量为750mg),收集所有的CMM在磁场帮助下乙醇洗涤以除去吸附的消旋物。洗脱清液也用HPLC在相同条件下分析。发现洗脱CMM 5次后,消旋物完全释放。得到的回收CMM在70℃真空干燥12h,得到738 mg CMM。
热重分析评估了Fe3O4@SiO2@CBDMP微球的有机层含量。 如fig4,与Fe3O4@SiO2相比Fe3O4@SiO2@CBDMPC微球在230℃有明显失重。可以推出Fe3O4@SiO2@CBDMPC微球在230℃和500℃表现高有机质量释放大约40%。有机物在500℃完全分解。
磁球磁性用振动样品磁强计(VSM)研究。 结果fig5,磁饱和值(Ms)的下降是由于TGA中高有机物含量造成的。 即,厚的非磁性有机CBDMPC层包覆在硅球表面,导致磁化强度降低。尽管Fe3O 4@SiO2@CBDMPC的Ms降低,但是在外加磁场下仍有强的磁响应,在60s内能完全分离。
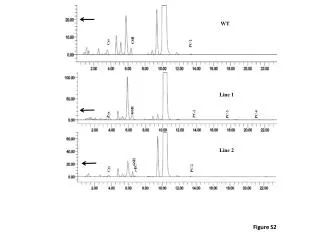
直接分离5种不同的消旋物研究CMM的手性识别能力,分离结果用自动数字旋光计分析。图table1.五种消旋物没有任何光学旋转,因为(+)(-)异构体产生等量相反光学旋光,导致消旋物的消除效应。当与CMM作用5min后,所有的5种消旋物表现正光学旋光。 可能原因是:纤维素衍生物具有三重螺旋的构型,因为其高度规律的螺旋结构导致其对消旋物的手性区分,还有氨基酸酯残基被认为是最重要的吸附位点。具有极性基团的消旋物可能主要通过氢键与氨基甲酸酯残基相互作用。消旋物和纤维素衍生物间的π-π相互作用在手性分离中也有重要作用。根据table1,当CMM在乙醇中用于直接手性分离时,主要通过其螺旋凹槽,氢键或π-π相互作用。
用NVM-GNPs检测cys的试验: 之前的工作情况 检测的cys的浓度范围 2——10 mg/L
近来工作进展: 由于NVM-GNPs稳定性的问题,如果GNPs稳定性非常好,在PBS中将对cys检测响应变差;而如果GNPs自身稳定性非常差,则在PBS中GNPs本身的酒红色就会因发生自团聚而逐渐变成蓝紫色。 [这时已经发生团聚了的GNPs不能用于cys的检测] 根据上次组会提出的问题,做了cys与GNPs反应随着时间变化的趋势: 检测加入0.4 mg/L cys后1h内A/A对时间的变化情况。 由图可以看出在低浓度的cys检测时,变色反应比较缓慢。 在加入cys之后,GNPs的溶液稍有颜色变化,从UV-vis谱上可以看出有些变宽,因此说明在此条件下,GNPs自身的稳定性仍然不太好。
线性范围:0.4 ——10 mg/L 线性范围:0.4 ——10 mg/L
20121018 线性 之前合成的NVM-GNPs储备液稀释备用 有时候NVM-GNPs稳定性非常好,如下情况:
20121019 线性 使用同一批的GNPs(比较稳定,所以想试试把GNPs的浓度稍微降低些,看看对于cys的检测,)使用的新配置的 50 mM pH=4.30 PBS,在这种情况下由于稀释了纳米粒子的浓度,NVM-GNPs自身的稳定性表现稍差,发现GNPs自身有团聚现象,反应放置1.5h立即测定UV-vis。平行做了两组实验。
20121025 干扰氨基酸 干扰氨基酸试验:
2012年10月31日 由于GNPs自身稳定性问题,有意注意合成条件。
2012年11月1日线性范围 水浴和室温条件下在低浓度的cys的检测线性都不好,NVM-GNPs稳定性较好的,可能需要更改PBS的离子强度和PH。
2012年11月5日 改变反应体系PBS做线性 由于之前使用的都是3mL 反应体系:2 mL 50 mM pH=4.45 PBS + 1 mL NVM-GNPs + 30 μL各级浓度的cys,室温条件下放置2h后测定UV-vis。但是,由于NVM-GNPs自身稳定性不好,或者是在50mM PBS中过于稳定,因而对于cys的检测效果都不好。 因此,考虑在使用稳定的NVM-GNPs的条件下,改变反应体系PBS的pH和浓度,以得到cys的线性范围。尝试了稳定的NVM-GNPs在100mM PBS中稳定性较好。如下图所示,在室温条件下与实验随行的空白NVM-GNPs在PBS中稳定性很好。 问题是最近做cys检测,在长波长处本来应该出现第二个吸收峰,现在变成了GNPs的吸收峰整体向长波长方向移动,而在做线性的图的时候,仍选择之前的几处波长的比值。
2012年11月6日 线性范围 100 mM 新PBS pH=4.59 合成的NVM-GNPs条件: 16.3 mg NVM + 200μL NaOH + 1mL 1% HAuCl4 75℃油浴2h。合成后GNPs在4℃保存老化一夜,然后离心浓缩保存,在100mM PBS中稳定性良好。
可能是反应时间不够2h,GNPs在较低浓度的cys的响应不成线性。可能是反应时间不够2h,GNPs在较低浓度的cys的响应不成线性。
选择线性范围比较好的一段: 范围0.06——4 mg/L 相关系数:0.9985
工作计划 针对实际样品检测问题 线性范围,不太稳定。