Download

1 / 39
1.29k likes | 3.95k Views
Platelet Disorders. Platelets. * Disc shape cells 2-4 mm in diameter. *They have blue grey cytoplasm with red lysosomal granules, but devoid of nuclei. *They are formed in BM from their progenitors the megakaryocytes .

E N D
Platelets *Disc shape cells 2-4 mm in diameter. *They have blue grey cytoplasm with red lysosomal granules, but devoid of nuclei. *They are formed in BM from their progenitors the megakaryocytes. *Megakaryocytes are giant cells with multilobed nuclei (4-6) that produce platelets from their cytoplasm by the process of budding.
*A hematopoietic growth factor for megakaryocytes is thrombopoietin *IL-11 also stimulates platelet production. *Each megakaryocyte produces 1000-3000 platelets. *Their normal count in peripheral blood is 150000-300000/µL. *One third resides in splenic pool & two thirds in circulation. *Their life span is 9-10 days.
Platelet function They contain 3 types of secretory granules: *lysosomes, *α-granules, *dense bodies.
α- granules contain *platelet-specific proteins (PF4, β-thromboglobulin) & *several growth factors including PDGF & TGF-β. *They also contain several hemostatic proteins including fibrinogen, FV, vWF synthesized by megakaryocytes.
Dense bodies (δ-granules) contain *ATP, *ADP, *Ca+2, *Serotonin
*Adhesion to blood vessel wall, *Aggregation to form the primary hemostatic plug at site of blood vessel injury, & * Release of vasoconstrictor substances from intracellular granules in response to variety of substances are the main functions of platelets. Platelets also provide surface for activation of coagulation factors. Activated platelets expose specific receptors that bind to F Xa & F Va increasing their local concentration thus accelerating prothrombin activation. F X is activated by F IXa & F VIII on platelet surface.
They contain a membrane - phospholipase C that can be stimulated by activating agents to hydrolyze endogenous phosphatidyl inositol to form a diglyceride. This is converted to arachidonic acid by diglyceride lipase enzyme. Arachidonic acid will be converted to prostaglandins (PG G2 & thromboxane A2) under the influence of cyclo-oxygenase .
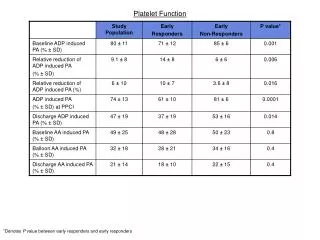
Platelet function tests 1. Bleeding time --- It is prolonged in thrombocytopenia, functional platelet abnormalities & vW disease. 2. Platelet aggregometry --- Response of platelets to variety of aggregating agents (ADP, fibrinogen, Collagen, ristocetin & adrenaline) is quantitated. It is useful in diagnosing hereditary platelet disorders.
Thrombocytopenia It is a condition in which platelet count is below normal level.
Causes 1- Aplastic anemia. 2- Leukemia. 3- Tumors metastatic to BM. 4- Myelofibrosis. 5- Megaloblastic anemia. 6- Peripheral platelet destruction a- Immune ITP, SLE, PAN, CLL, NHL, infectious mono, CMV infection, HIV, quinine, quinidine, heparin & sulfa. b- Non-immune DIC, TTP, PNH, acute transplant rejection. 7- Disorders of distribution--- Hypersplenism. 8- Dilutional --- Old banked blood transfusion.
Idiopathic Thrombocytopenic Purpura: (ITP) It is a bleeding disorder caused by autoimmune Abs destroying patient's own platelets by phagocytosis in the spleen by Mφ (to a lesser degree in the liver).
Epidemiology *Children are affected by an acute type of illness following URT infection (usually viral). *♂ = ♀ incidence. *Adults are affected by a more gradual onset of disease with chronic course & without preceding illness. *Age < 40 years *♀: ♂ = 3-4:1
S & S Skin shows petechiae (pinpoint red hemorrhagic spots) & ecchymoses. Mucosal bleeding --- bleeding gum, epistaxis, menorrhagia, metrorrhagia, GI bleeding & hematuria Intracranial hemorrhage is seen in 1%. Risk of death 5% No splenomegaly.
Dx Platelet count < normal (Thrombocytopenia) * If > 100000/µL no spontaneous bleeding even with major surgery *If 50000-100000/µL Bleeding more than normal with severe trauma *If 20000-50000/µL Bleeding occurs with minor trauma *If < 20000/µL spontaneous bleeding occurs *If < 10000/µL patients are at high risk of severe bleeding
Blood film shows scanty platelets BM examination is normal with abundant megakaryocytes Serum antiplatelet Abs --- anti-Gp IIb-IIIa Platelet surface attached Abs assay (IgG 90%) – more complex & its significance is not clear
DDx SLE, PAN, CLL, NHL, EBV & CMV mononucleosis, HIV
℞ Children have a self-limited disease & 70% recover in 4-6 weeks. Asymptomatic adult pts with platelet count > 40000/µL are observed periodically & need no ttt
Prednisolone is the 1st line of ttt 1-2 mg/kg body wt daily * inhibits Mφ ingestion of Ab-coated platelets * suppresses Ab synthesis * have stabilizing effect on blood vessels wall. -- 80-90% of pts will have rise of platelet count to hemostatic level within 2-3 weeks -- Tapered slowly after achieving normal count
Splenectomy * improves the count in 70% pts & sustained remission in 60% * Up to 2 weeks later * For pts not responding to steroids * No test can predict which pts can detect which pts respond to it
Intravenous Immunoglobulin (IVIG) * Effective in pts with active bleeding & before major surgery * Rise platelet count in 3-5 days (The most rapid agent) * Blocks receptors to Abs on Mφ therefore inhibiting phagocytosis * High dose needed 1g/kg B wt/day on 2 successive days * Very expensive * Its effect is unfortunately transient
Anti-Rh antibody (RhoGAM) * Less expensive alternative to IVIG * Mechanism is the same i.e. reticuloendothelial cells blockade
Danazol (Danol) * 200mg three times per day * Induces remission in 40% of cases * Response is delayed 4-6 weeks * Impeded androgen * Mechanism of action remains unknown
Vincristine (Oncovin) & Vinblastine (Velbe) * Cytotoxic drugs * Given IV * Raise platelet within 1-2 weeks * Transient & nonsustained response
Immunosuppressants Cyclophosphamide & Azathioprine * Improve platelet count in 20-30% of cases * Risk of toxicity has to be weighed against potential benefit
Refractory cases – Combination chemotherapy -- Pulsed high dose dexamethasone
ITP in Pregnancy * Complicated by additional risk to the fetus (thrombocytopenia due to maternal Abs) , newborn intraventricular hemorrhage & GI bleeding * C/S delivery is recommended to decrease risk of intracranial bleeding in the newborn.
Henoch-Schonlein Purpura Also called anaphylactoid purpura Symmetrical palpable purpura is seen on the extensor surfaces of the limbs & buttocks (maculopapular rash). It is usually seen in children (rare in adults)
Patients may give a history of a recent infection Manifestations in addition to palpable purpura include arthralgias, abdominal pain& melena Complications include glomerulonephritis, hypertension & intussusception No splenomegaly Platelet count is normal
Histopathologic examination shows small blood vessel leucocytoclastic vasculitis with IgA & complement deposition It is self limited over 1-2 months Symptomatic response is seen with Prednisolone & analgesics Relapses may complicate the course of the disease Bad prognostic factors – Adult age, renal involvement & recurrent relapsing type
Functional platelet disorders Hereditary • Glanzmann's thrombasthenia • Bernard-Soulier syndrome • Storage pool disease
Glanzmann's thrombasthenia • AR inheritance • Prolonged bleeding time • Platelets fail to aggregate with ADP, adrenaline, collagen, or thrombin • Deficiency of platelet membrane GP IIb-IIIa (two glycoproteins that normally serve as the receptor for fibrinogen in activated platelets)
Bernard-Soulier syndrome • AR inheritance • Deficiency of platelet membrane GPIb-IX causing defective binding of vWF • Peripheral blood smear – Giant platelets • Platelet aggregate normally with ADP, collagen, or adrenaline, but fail to respond to ristocetin
Storage pool disease • AD inheritance • Platelet storage granules are decreased in no. & content • Abnormal granules formation in megakaryocytes • Mild bleeding mainly in women • Decreased platelet aggregation because of inadequate secretion of ADP • Sometimes associated with oculocutaneous albinism,