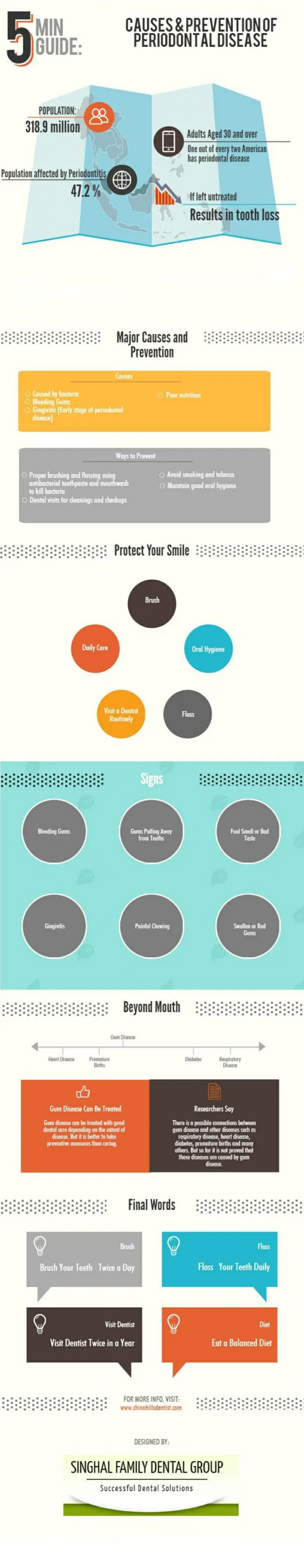
Download

1 / 41
410 likes | 523 Views
P. St George-Hyslop Centre for Research in Neurodegenerative Diseases, Toronto Western Hospital Research Institute, University of Toronto, Toronto, Ontario, CANADA. Insights into Basic and Clinical Neurobiology Derived from the Analysis of Genetic causes of Neurodegenerative Disease.

E N D
P. St George-Hyslop Centre for Research in Neurodegenerative Diseases, Toronto Western Hospital Research Institute, University of Toronto, Toronto, Ontario, CANADA Insights into Basic and Clinical Neurobiology Derived from the Analysis of Genetic causes of Neurodegenerative Disease
Overview • Genetics and Biology of Dementias • Alzheimer Disease: • APP, PS1, PS2, APOE ε4 • Other unidentified genes • Fronto-temporal Dementia (& PSP , CBD) • Tau • Dementia with Lewy Bodies • APOE ε4 • Current knowledge of known disease causing pathways; • Application of current knowledge • Prediction of future risks, pharmacogenomics • Design of rational therapeutics
Emerging Concept:neurotoxic intra- or extra-cellular deposition of insoluble proteins (-sheet conformation) is the cause of many neurodegenerative diseases • Disease Protein Enabling event • Alzheimer Disease Aβ (βAPP) β- /γ-secretase • Frontotemporal Dementia Tau ? • Creutzfeldt-Jacob PrPSc (PrPc) ? • Familial Encephalopathy Neuroserpin ? • Familial British Dementia ABri (BRI) Furin cleavage • Parkinson’s Disease α-synuclein ?
What causes Alzheimer Disease? • Genetic Factors (40% of attributable population risk): • Mutations in genes: • Amyloid Precursor Protein (APP); • Presenilin 1 (PS1); • Presenilin 2 (PS2); • Apolipoprotein E (APOE ε4); • Other genes on other chromosomes. • Environmental Factors (± genetic predispositions): • Evidence for specific environmental factors is not robust • Lower childhood education • Head Injury • Cerebrovascular disease • ?Aluminium
Genetic and “non-genetic” cases are indistinguishable • Genetic and non-genetic cases have identical: • Clinical features; • Brain pathology; • Brain biochemistry (increased brain levels of Amyloid β-peptide (Aβ) and tau); • Mortality.
Genetic Determinants of Alzheimer’s Disease PresenileFamilial AD SenileFamilial AD SporadicAD Presenilin 2gene(chr 1) 45–84 yrs Presenilin 1gene(chr 14) age: 25–60 yrs APPgene(chr 21) 40–65 yrs APOE 4 allele(chr 19) >50 yrs Other genes yet to be identified
The APP gene encodes a Type 1 membrane protien, a fragment of which accumulates in AD brain Citron et al. Nature Med.3: 67-72, 1997 Aβ peptide domain APP Cell membrane
Aβ40 >> Aβ42 AICD (?Signalling) Physiological Endo-proteolytic Processing of APP Citron et al. Nature Med.3: 67-72, 1997 Pardossi-Piquard R et al. Neuron46:541-554, 2005. Uptake, chaperoning, & degradation of Aβ by neprilysin, IDE, others b-secretase g-secretase b a g APP Transcriptional induction Cell membrane a-secretase
Aβ Extracellular TM domain Intracellular AICD (?Signalling) Mutations Causing Alzheimer Disease cause mis-processing of APP Citron et al. Nature Med.3: 67-72, 1997 APP mutations Uptake, chaperoning, & degradation of Aβ by neprilysin, IDE, others g-secretase b-secretase b a g APP a-secretase
Extracellular TM domain Intracellular b a g APP FAD-causing mutations in APP are localized in/around the Aβpeptide domain. • Codon Mutation Phenotype Effect • 670/671 Lys-Met/ FAD β-secretase cleavage Asn-Leu • 692 Ala->Gly FAD Fibrillogenesis/toxicity • 693 Glu->Gln Haemorrhage Fibrillogenesis/toxicity Glu->Gly Haemorrhage Fibrillogenesis/toxicity • 694 Asp->Asn Haemorrhage Fibrillogenesis/toxicity • 713 Ala->Thr FAD • 714 Thr->Ile FAD N-truncated Aβ42 • 715 Val->Met FAD N-truncated Aβ42 • 716 Ile->Val FAD Aβ42 • Val->Ile/Phe FAD Aβ42 • /Gly • 723 Leu->Pro FAD Aβ42
FAD-causing mutations in APP are alter the amount or the fibrillogenic potential of Aβpeptide • Codon Mutation Phenotype Effect • 670/671 Lys-Met/ FAD β-secretase cleavage Asn-Leu • 692 Ala->Gly FAD Fibrillogenesis/toxicity • 693 Glu->Gln Haemorrhage Fibrillogenesis/toxicity Glu->Gly Haemorrhage Fibrillogenesis/toxicity • 694 Asp->Asn Haemorrhage Fibrillogenesis/toxicity • 713 Ala->Thr FAD • 714 Thr->Ile FAD N-truncated Aβ42 • 715 Val->Met FAD N-truncated Aβ42 • 716 Ile->Val FAD Aβ42 • Val->Ile/Phe FAD Aβ42 • /Gly • 723 Leu->Pro FAD Aβ42
Ab Extracellular TM domain Intracellular AICD (?Signalling) Mutations Causing Alzheimer Disease cause mis-processing of APP Citron et al. Nature Med.3: 67-72, 1997 APP mutations PS1/PS2 mutations Uptake, chaperoning, & degradation of Aβ by neprilysin, IDE, others g-secretase b-secretase b a g APP a-secretase
>100 missense/in-frame splicing mutations in PS1 scattered throughout PS1 molecule; • > 12 mutations in PS2; • Mutations in PS1 and PS2 often affect orthologous residues. Cytoplasm Membrane Lumen Naturally Occurring Mutations in Presenilins Alter APP Processing • Predicted to encode homologous polytopic transmembrane proteins (PS1 and PS2). • Contain conserved aspartate residues in transmembrane domains (protease active site). XD XGXGD • PS1 and PS2 mutations all alter Aβ production – increase Aβ42. Sherrington et al. Nature375: 754-760, 1995 Rogaev et al Nature376: 775-778, 1995 Citron et al. Nature Med.3: 67-72, 1997
APH-1 Nicastrin Ab Presenilin PEN-2 Presenilin Proteins Form a Complex With Nicastrin APH-1 and PEN-2 To Cleave Amyloid Precursor Protein (APP) and generate neurotoxic Aβ peptide. AICD Golgi/ER ε-site Cytoplasm D D Membrane Lumen/ Cell surface g-site Alzheimer Disease _ • Similar presenilin-dependent • intramembranous cleavages for: • Notch • Delta • p75 • LRP1 • SorLA • Others... Sherrington, Nature, 1995 Rogaev, Nature, 1995 Katayama, Nature Cell Biol, 1999 Yu, Nature, 2000 Chen, Nature Cell Biol, 2002 Sisodia, Nature Neurosci, 2002 Pardossi-Piquard Neuron, 2005
Ab42 Extracellular TM domain Intracellular AICD (?Signalling) Presenilin Mutations Cause Alzheimer Disease by altering γ-secretase cleavage of APP Citron et al. Nature Med.3: 67-72, 1997 APP mutations PS1/PS2 mutations Uptake, chaperoning, & degradation of Aβ by neprilysin, IDE, others g-secretase-42 b-secretase b a g APP a-secretase
Apolipoprotein E and Alzheimer’s Disease • APOE has 3 variants: 2, 3, 4; • APOE 2 increased frequency in normal elderly, reduced frequency in AD; • APOE 4 associated with Sporadic/familial AD (dose-dependent relationship with age of onset); • APOE 4 association not specific to AD, and not all APOE 4 carriers will succumb to disease. • APOE ε4 appears to block removal of Aβ via LRP receptors, causing accumulation of Aβ.
Ab Extracellular TM domain Intracellular AICD (?Signalling) Ab accumulates Ab aggregates into neurotoxic protofibrils Mutations Causing Alzheimer Disease cause mis-processing of APP Citron et al. Nature Med.3: 67-72, 1997 APP mutations PS1/PS2 mutations APOE e4 g-secretase b-secretase ↓Uptake, chaperoning, & degradation of Aβ X b a g APP a-secretase
Enhancer and suppressor interactions amongst genes causing Alzheimer Disease Gene interactions in human patients with AD: • APP717 mutation + APOE e4 allele = earlier onset (enhancer); • APP717 mutation + APOE e2 allele = delayed onset (suppressor); • PS1E280A + APOE ε4 = earlier disease (enhancer) • PS2N141V + APOE ε4 = earlier disease (enhancer) . Gene interactions In animal models • APP717 mutation + PS10/0 = no disease (suppressor); • APP717 mutation + PS1mutations = enhanced disease (enhancer). St George-Hyslop et al Science263:536-537, 1994 Pastor, P. et al. Ann Neurol54, 163-9 (2003)
Suppressor APP genotype (A= APP717) APOE Genotype WT/WT A717/WT ε2/ε3 ε3/ε3 A717/WT A717/WT A717/WT ε2/ε3 ε4/ε3 ε4/ε3 APPV717I + APOE ε2 carrier eventually developed AD, but at >2 SD beyond mean age-of-onset. Elderly (>65yrs old) asymptomatic carrier of APPV717I mutation
Enhancer and suppressor interactions amongst genes causing Alzheimer Disease Gene interactions in human patients with AD: • APP717 mutation + APOE e4 allele = earlier onset (enhancer); • APP717 mutation + APOE e2 allele = delayed onset (suppressor); • PS1E280A + APOE ε4 = earlier disease (enhancer) • PS2N141V + APOE ε4 = earlier disease (enhancer) . Gene interactions In animal models • APP717 mutation + PS10/0 = no disease (suppressor); • APP717 mutation + PS1mutations = enhanced disease (enhancer). St George-Hyslop et al Science263:536-537, 1994 Pastor, P. et al. Ann Neurol54, 163-9 (2003)
APP x PS1 mice - 2 months Enhancer effect of cross-breeding mutant PS1 and mutant APP mice APP mice – 2 months PS1 mice - 2 months
Enhancer and suppressor interactions amongst genes causing Alzheimer Disease • Confirms that the known AD genes really do act in the same biochemical pathway affecting APP processing. St George-Hyslop et al Science263:536-537, 1994 Pastor, P. et al. Ann Neurol54, 163-9 (2003)
General Paradigms for Gene Discovery LINKAGE BASED CASE : CONTROL ASSOCIATION • Easy to collect sporadic cases • Cheap, quick • Easy to mess up • Requires assumption that cases and controls are from same founder population.. • Difficult to collect families • Expensive • Relatively few assumptions • Robust directly observable results
What are the other AD genes? Case:Control > 100 candidate genes reported to be associated with AD; Generally had poor track-record of replication (NB: one or two ‘independent replications’ in the face of many non-replications = non-replication); Family linkage-based method Confirmed localization of an AD-gene to broad region of chromosome 10 containing several hundred genes (the specific gene remains to be found); Confirmed localization of an AD-gene to broad region of chromosome 12 containing several hundred genes (the specific gene remains to be found)
What is the role for themicrotubule associated protein Tau and neurofibrillary tangles?
Fronto-temporal dementia:molecular genetics • Mutations in Tau gene on chromosome 17q in ~10-40% of FTD cases; • Mutations disturb binding of tau protein to microtubules, causing accumulation of free unbound tau; • Free unbound tau aggregates into fibrils and these then coalesce into paired helical filaments as the neurofibrillary tangle; • The tau fibrils then injure cells (but mechanism is unclear).
Conclusions to Be Drawn From the Discovery of Pathogenic Mutations in Tau in FTD • Disturbed tau/microtubule homeostasis, regardless of cause, is toxic to neurons
Neuronal injury Ab accumulation initiates a biochemical cascade leading to neuronal death Dementia Cause: (eg gene defect) Ab peptide accumulation Neuronal dysfunction and death Altered Tau metabolism
How is this knowledge applied for patients? • Adjunctive Diagnostics • Therapeutic Targets
Prediction of future risk for AD? • Testing and genetic counselling feasible for: • Highly penetrant forms, with • Clear patterns of inheritance, and • Relatively predictable age-of-onset: • PS1 • APP • Tau • Testing and genetic counselling not presently feasible/useful for: • Incompletely penetrant forms with variable age-of-onset: • PS2 • APOE • Putative genes on chromosomes 10, 12 etc • NB: Advent of future therapies may make even fuzzy-risk data from such genes useful
Can Genetics Predict Conversion From MCI To AD? • Intuitive expectation: • Carrier of AD risk allele with MCI would be more likely to convert to AD. • Actual data available only for ApoE • ApoE ε4 predictive: • Petersen et al, JAMA 274: 538,1995 • Bartrez-Faz et al, JAGS 49: 485, 2001 • ApoE ε4 not predictive: • Marquis et al, Arch. Neurol. 59: 601, 2002 • Tierney et al, Neurol. 46: 149, 1996.
Gene 1 Environment factor 1 Gene 2 Rx 2 Rx 1 Prediction of therapeutic response • Theoretically reasonable; • Remains to be validated. AD Step 2 Step 4 Step 1 Step 3
Neuronal injury Using Ab accumulation pathways as a target for therapies Dementia Cause: (eg gene defect) Ab peptide accumulation Neuronal dysfunction and death Altered Tau metabolism
Neuronal injury Exploiting Knowledge Gained to Create New Diagnostics and Therapeutics • Anti-Ab antibodies to remove Ab; • Block enzymes; • Block aggregation. Dementia X Cause: (eg gene defect) Ab peptide accumulation Neuronal dysfunction and death Altered Tau metabolism Janus et al Nature. 408: 979-982, 2000, McLaurin et al, Nature submitted, 2004
Ab AICD (?Signalling) Ab accumulates Ab aggregates into neurotoxic protofibrils How can the amyloid cascade be blocked? Citron et al. Nature Med.3: 67-72, 1997 Pharma: Vaccine: toxic Pharma: Uptake, chaperone, or degradation (by neprilysin). g-secretase b-secretase Pharma b a g X APP Cell membrane
Conclusions: • All known genes causing AD modulate APP and Aβ processing; • Neurodegeneration from mutations in tau prove that tau accumulation is also a toxic event (regardless of whether caused by mutation in tau or due to Aβ accumulation) • Knowledge of pathway will provide targets for disease-modifying therapies.
Acknowledgements S. Arawaka F. Chen L. Farrer, P. Fraser YJ. Gu H. Hasegawa M. Ikeda T. Katayama T. Kawarai G. Levesque M. Nishimura A. Petit E. Rogaeva N. Sanjo P. St George-Hyslop D. Westaway A. Bruni, F. Checler JF Foncin, G. Marcon, M. Mortilla, A. Orlacchio, E. Paitel S. Piacentini, L. Pinessi, I. Rainero, S. Sorbi, R. Tupler, G. Vaula Canadian Institutes of Health Research Howard Hughes Medical Institute Alzheimer Society of Ontario, Canadian Genetic Diseases Network
CONTACT INFORMATION • Analysis of familial cases: P. St George-Hyslop, University of Toronto tel: 416-978-7460 p.hyslop@utoronto.ca • Animal models (transgenic mice etc): David Westaway David.westaway@utoronto.ca • Reagents (clones, cell lines, antibodies, etc) P. St George-Hyslop, University of Toronto p.hyslop@utoronto.ca