Download

1 / 88
1.02k likes | 1.46k Views
Protein Structure. Lesk, chapter 5 Details on SCOP and CATH can be found in Structural Bioinformatics, Bourne/Weissig, chapter 12 and 13. Folding. Proteins are linear polymer mainchains with different amino acid side chains Proteins fold spontaneously reaching a state of minimal energy

E N D
Protein Structure Lesk, chapter 5 Details on SCOP and CATH can be found in Structural Bioinformatics, Bourne/Weissig, chapter 12 and 13
Folding • Proteins are linear polymer mainchains with different amino acid side chains • Proteins fold spontaneously reaching a state of minimal energy • Side and main chains interact with one another and with solvent • Example movie Jones, D.T. (1997) Successful ab initio prediction of the tertiary structure of NK-Lysin using multiple sequences and recognized supersecondary structural motifs. PROTEINS. Suppl. 1, 185-191
Examining Proteins • Specialised tools with different views of structure • Corey, Pauling, Koltun (CPK) • Diameter of sphere ~ atomic radius • Hydrogen white, carbon grey, nitrogen blue, oxygen red, sulphur yellow • Cartoon • Wire • Balls
Residue Protein Folding • Conformation of residue • Rotation around N-Cabond, (phi) • Rotation around Ca-C bond, (psi) • Rotation around peptide bond (omega) • Peptide bond tends to be • planar and • in one of two states: • trans180 (usually) and • cis, 0(rarely, and mostly proline) Image taken from www.expasy.org/swissmod/course
Sasisekharan-Ramakrishnan-Ramachandran plot • Solid line = energetically preferred • Outside dotted line = disallowed • Most amino acids fall into R region (right-handed alpha helix) or -region (beta-strand) • Glycine has additional conformations (e.g. left-handed alpha helix = L region) and in lower right panel Image taken from www.expasy.org/swissmod/course
Ramachandran plot Plot for a protein with mostly beta-sheets Example for conformations Image taken from www.expasy.org/swissmod/course
Helices and Strands • Consecutive residues in alpha or beta conformation generate alpha-helices and beta-strands, respectively • Such secondary structure elements are stabilised by weak hydrogen bonds • They are by turns or loops, regions in which the chain alters direction • Turns are often surface exposed and tend to contain charged or polar residues
Alpha Helix • Residue j is hydrogen-bonded to residue j+4 • 3.6 residues per turn • 1.5A rise per turn • Repeat every 3.6*1.5A = 5.4 A • = -60 , = -45 Image taken from www.expasy.org/swissmod/course
Beta strand Image taken from www.expasy.org/swissmod/course
Beta Sheets Image taken from www.expasy.org/swissmod/course
Turn • Residue j is bonded to residue j+3 • Often proline and glycine Image taken from www.expasy.org/swissmod/course
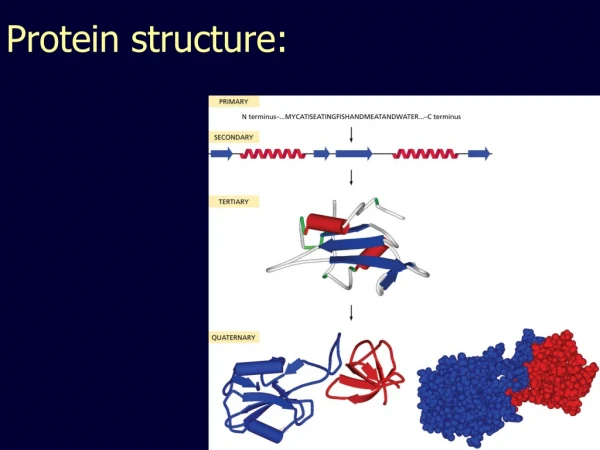
How to Fold a Structure • All residues must have stereochemically allowed conformations • Buried polar atoms must be hydrogen-bonded • If a few are missed, it might be energetically preferable to bond these to solvent • Enough hydrophobic surface must be buried and interior must be sufficiently densely packed • There is evidence, that folding occurs hierarchically: First secondary structure elements, then super-secondary,… • This justifies hierarchic approach when simulating folding
Structure Alignment + Slides from Hanekamp, University of Wyoming, www.uwyo.edu
Structure Alignment • In the same way that we align sequences, we wish to align structure • Let’s start simple: How to score an alignment • Sequences: E.g. percentage of matching residues • Structure: rmsd (root mean square deviation)
a b Root Mean Square Deviation • What is the distance between two points a with coordinates xa and ya and b with coordinates xb and yb? • Euclidean distance:d(a,b) = √(xa--xb )2 + (ya -yb )2 + (za -zb )2
Root Mean Square Deviation • In a structure alignment the score measures how far the aligned atoms are from each other on average • Given the distances di between n aligned atoms, the root mean square deviation is defined as rmsd = √ 1/n ∑ di2
Quality of Alignment and Example • Unit of RMSD => e.g. Ångstroms • Identical structures => RMSD = “0” • Similar structures => RMSD is small (1 – 3 Å) • Distant structures => RMSD > 3 Å • Structural superposition of gamma-chymotrypsin and Staphylococcus aureus epidermolytic toxin A
Pitfalls of RMSD • all atoms are treated equally (e.g. residues on the surface have a higher degree of freedom than those in the core) • best alignment does not always mean minimal RMSD • significance of RMSD is size dependent From www.uwyo.edu/molecbio/LectureNotes/ MOLB5650
Alternative RSMDs • aRMSD = best root-mean-square deviation calculated over all aligned alpha-carbon atoms • bRMSD = the RMSD over the highest scoring residue pairs • wRMSD = weighted RMSD Source: W. Taylor(1999), Protein Science, 8: 654-665. http://www.prosci.uci.edu/Articles/Vol8/issue3/8272/8272.html#relat From www.uwyo.edu/molecbio/LectureNotes/ MOLB5650
Computing Structural Alignments • DALI (Distance-matrix-ALIgnment) is one of the first tools for structural alignment • How does it work? • Atoms: • Given two structures’ atomic coordinates • Compute two distance matrices: • Compute for each structure all pairwise inter-atom distances. • This step is done as the computed distances are independent of a coordinate system • The two original atomic coordinate sets cannot be compared, the two distance matrices can • Align two distance matrices: • Find small (e.g. 6x6) sub-matrices along diagonal that match • Extend these matches to form overall alignment • This method is a bit similar to how BLAST works. • SSAP (double dynamic programming) in term 3.
DALI Example • The regions of common fold, as determined by the program DALI by L. Holm and C. Sander, in the TIM-barrel proteins mouse adenosine deaminase [1fkx] (black) and Pseudomonas diminuta phosphotriesterase [1pta] (red):
Protein zinc finger (4znf) Slides from Hanekamp, University of Wyoming, www.uwyo.edu
Superimposed 3znf and 4znf 30 CA atoms RMS = 0.70Å 248 atoms RMS = 1.42Å Lys30 Slides from Hanekamp, University of Wyoming, www.uwyo.edu
Superimposed 3znf and 4znf backbones 30 CA atoms RMS = 0.70Å Slides from Hanekamp, University of Wyoming, www.uwyo.edu
RMSD vs. Sequence Similarity • At low sequence identity, good structural alignments possible Picture from www.jenner.ac.uk/YBF/DanielleTalbot.ppt
Why classify structures? • Structure similarity is good indicator for homology, therefore classify structures • Classification at different levels • Similar general folding patterns (structures not necessarily related) • Possibly low sequence similarity, but similar structure and function implies very likely homology • High sequence similarity implies similar structures and homology • Classification can be used to investigate evolutionary relationships and possibly infer function
Structure Classification • SCOP: Structural Classification of Proteins • Hand curated (Alexei Murzin, Cambridge) with some automation • CATH: Class, Architecture, Topology, Homology • Automated, where possible, some checks by hand • FSSP: Fold classification based on Structure-Structure alignment of Proteins • Fully automated • Reasonable correspondance (>80%)
Evolutionary Relation • Strong sequence similarity is assumed to be sufficient to infer homology • Close structural and functional similarity together are also considered sufficient to infer homology • Similar structure alone not sufficient, as proteins may have converged on structure due to physiochemical necessity • Similar function alone not sufficient, as proteins may have developed it due to functional selection • In general, structure is more conserved than sequence • Beware: Descendents of ancestor may have different function, structure, and sequence! Difficult to detect
What is a domain? • Functional: Domain is “independent” functional unit, which occurs in more than one protein • Physiochemical: Domain has a hydrophobic core • Topological: Intra-domain distances of atoms are minimal, Inter-domain distances maximal • Difficult to exactly define domain • Difficult to agree on exact domain border
Domains re-occur • A domain re-occurs in different structures and possibly in the context of different other domains • P-loop domain in • 1goj: Structure Of A Fast Kinesin: Implications For ATPase Mechanism and Interactions With MicrotubulesMotor Protein (single domain) • 1ii6: Crystal Structure Of The Mitotic Kinesin Eg5 In Complex With Mg-ADPCell Cycle (two domains)
Domains re-occur 1in5: interaction of P-loop domain (green & orange) and winged helix DNA binding domain 1a5t: interaction of P-loop domain (green & orange) and DNA polymerase III domain
Domains have hydrophobic core Ala: 1.800 Arg: -4.500 Asn: -3.500 Asp: -3.500 Cys: 2.500 Gln: -3.500 Glu: -3.500 Gly: -0.400 His: -3.200 Ile: 4.500 Leu: 3.800 Lys: -3.900 Met: 1.900 Phe: 2.800 Pro: -1.600 Ser: -0.800 Thr: -0.700 Trp: -0.900 Tyr: -1.300 Val: 4.200 • Kyte J., Doolittle R.F, J. Mol. Biol. 157:105-132(1982).
Intra-domain distances minimal • Distances between atoms within domain are minimal • Distances between atoms of two different domains are maximal
PDB, Proteins, and Domains • Ca. 20.000 structures in PDB • 50% single domain • 50% multiple domain • 90% have less than 5 domains
1AON, Asymmetric Chaperonin Complex Groel/Groes/(ADP)7 A structure with 49 domains
Alpha+Beta (279) All alpha (218) All Beta (144) Alpha/Beta (136) Immunoglobulin-like (23) Trypsin-like serine proteases (1) Transglutaminase (1) Immunoglobulin (6) C1 set domains (antibody constant) V set domains (antibody variable) SCOP: Structural Classification of Proteins top CLASS FOLD SUPERFAMILY FAMILY
Class • All beta • (possibly small alpha adornments) • All alpha • (possibly small beta adornments)
Class • Alpha/beta (alpha and beta) = single beta sheet with alpha helices joining C-terminus of one strand to the N-terminus of the next • subclass: beta sheet forming barrel surrounded by alpha helices • sublass: central planar beta sheet • Alpha+beta (alpha plus beta) = Alpha and beta units are largely separated • Strands joined by hairpins leading to antiparallel sheets
Class • Multi-domain proteins • have domains placed in different classes • domains have not been observed elsewhere • E.g. 1hle
Class • Membrane (few and most unique) and cell surface proteins • E.g. Aquaporin 1ih5
Small Proteins E.g. Insulin, 1pid Class
Class • Coiled coil proteins • E.g. 1i4d, Arfaptin-Rac binding fragment
Class • Low-resolution structures, peptides, designed proteins • E.g. 1cis, a designed protein, hybrid protein between chymotrypsin inhibitor CI-2 and helix E from subtilisin Carlsberg from Barley (Hordeum vulgare), hiproly strain
Fold, Superfamily, Family • Fold • Common core structure • i.e. same secondary structure elements in the same arrangement with the same topological structure • Superfamily • Very similar structure and function • Family • Sequence identity (>30%) or extremely similar structure and function
Uses of SCOP • Automatic classification • Understanding of protein enzymatic function • Use superfamily and fold to study distantly related proteins • Study sequence and structure variability • Derive substitution matrices for sequence comparison • Extract structural principles for design • Study decomposition of multi domain proteins • Estimate total number of folds • Derived databases