Download

1 / 28
280 likes | 316 Views
Genome Characterization. Assembly/sequencing. Assigned reading: Ch 9. BIO520 Bioinformatics Jim Lund. The (original) genome sequencing process. Organism Selection. Library Creation. Sequencing. Assembly. Gap Closure. Finishing. Annotation. The (current) genome sequencing process.

E N D
Genome Characterization Assembly/sequencing Assigned reading: Ch 9 BIO520 Bioinformatics Jim Lund
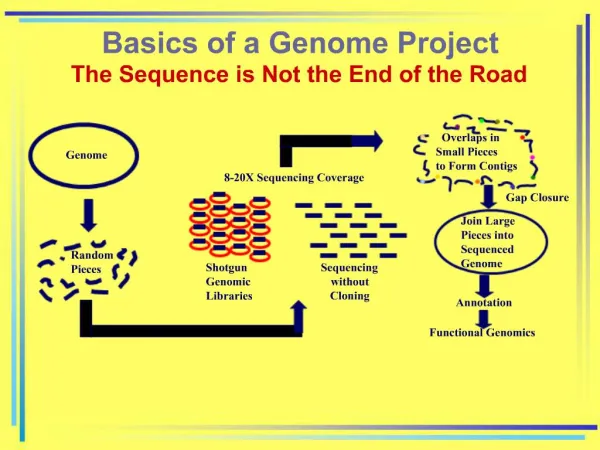
The (original) genome sequencing process Organism Selection Library Creation Sequencing Assembly Gap Closure Finishing Annotation
The (current) genome sequencing process Organism Selection Next gen. random sequencing lets library generation get skipped Sequencing Assembly Gap closure and finishing often get skipped, at least for now. Annotation
Contigs, Islands contigs Island
Assembly pipeline • Sequence reads. • Phred: base calling. • crossmatch: screen out vector, E. coli sequence. • Phrap: assemble contigs. • Consed: view assembly, correct problems. • Finishing.
Assembly Methods • Strip out vector (or contaminant) • Mask known repeats • Trim off unreliable data • Find Matches (n seq x n seq comparisons) • how long (what ktuple [10 common]) • how perfect (reliability index) • where to look? (ends only vs entire)
Assembly Programs • PHRAP FAMILY • phred/phrap/consed/cross_match • Developed by Phil Green, U of Wash. • Other assemblers • phrap, kangaroo, phrapo, • CAP, TIGRAssembler,... http://www.phrap.org/
Assembly • Phred -reads DNA sequencing trace files, calls bases, and assigns a quality value to each called base. • The quality value is a log-transformed error probability, specifically: Q = -10 log10( Pe ) • Q = quality value, Pe = error probability. • Q= 20 -> 1% chance of miscall, Q= 30 -> 0.1% chance of miscall. • Phrap -assembles shotgun DNA sequence data. • Consed/Autofinish -view, edit, and finish sequence assemblies created with phrap. • Allows the user to pick primers and templates • Suggests additional sequencing reactions • Suggest digests and forward/reverse pair information to check accuracy of assembly.
Poisson statistics for sequencing completion L=read length N=#reads G=genome size P0=e-L(N)/G Coverage 1 = 1-fold = 1X 1 3 8 10 50 % not sequenced 37 5 0.03 0.005 < 1e20 E. coli 15kb H. sapiens 900kb
Gaps Number of Gaps = Ne-c 150kb Target Clone, 500 bp reads N=# of reads c = fold coverage Coverage, reads 1, 300 5, 1500 8, 2400 10, 3000 50, 15000 Gaps 111 10 1 0 0
Gaps Number of Gaps = Ne-c Human genome, 3Gb, 1,000 bp reads N=# of reads c = fold coverage Coverage, reads 1, 3e6 5, 1.5e7 8, 2.4e7 10, 3e7 50, 3.75e8 Gaps 1,000,000 100,000 8,000 1,400 7 454 Seq, 400bp reads
Contigs, Islands C T T T contigs HTGS Island Finish!!
Finishing • GOALS • >95% coverage on BOTH strands • every base covered 3X • resolve ambiguities • Finish when random no longer productive (~8X range)
Sequence finishing. How? • Identify gaps, ambiguities • Captured gaps: gaps is contained in a clone • Extend from end of contigs • Resequencing, new chemistry. • Specific primers • Subcloning and sequencing. • Uncaptured gaps. • New specific primers • PCR across gap, sequence PCR product. • Resolve ambiguities • Consensus or resequence • Specific primers, different chemistry
Large clone sequencing process • Phase 1: Unfinished, may be unordered/unoriented contigs, with gaps. • Phase 2: Unfinished, fully oriented and ordered sequence, may contain gaps and low quality sequences • Phase 3: Finished, no gaps.
Genome assembly after initial contigs are made • Order clones/contig sequences: • Sequence overlaps. • Clone/contig end sequences. • Clone fingerprints. • Anchor using other maps • Sequence based markers on genetic or physical maps. • Conserved synteny to other genomes. • Easiest when re-sequencing, e.g, another human genome!
Process Control • LIMS • Laboratory information management system • AIMS • Analysis information management system
Hard genome sequencing problems • Repeats • Complex genome structures Where does a clone from a repetitive region map?
Approaches to sequencerepeat problems • Multiple fragment sizes in 1 project • Use length/distance info • New assemblers, eg. ARACHNE
Results of Multi-length Fragment Assembly • Contigs • “Supercontigs” • Clone links for finishing • Clone map
DOE Joint Genome Institute (JGI) Prokaryote Finishing Standards • All low-quality areas (<Q30) are reviewed and resequenced. • The final error rate must be less than 0.2 per 10 Kb. • No single-clone coverage is permitted (minimum of 2x depth everywhere). • Single-stranded regions are manually inspected and quantified. • All positions where an aligned high-quality read (>Q29) disagrees with the consensus base are checked. • All strings of xxxx are resolved in the final sequence. • All repeats are verified. • The ends of final contigs (chromosomes, plasmids) are checked • The final assembly is given a manual QC check.
Completed genomes Eremothecium gossypii Homo sapiens Kluyveromyces lactis Leishmzania major Friedlin Mus musculus Oryza sativa Saccharomyces cerevisiae Schizosaccharomyces pombe Trypanosoma cruzi Yarrowia lipolytica 23 complete, 329 in assembly, in progress 389 Arabidopsis thaliana Caenorhabditis elegans Candida glabrata Cryptococcus neoformans Cyanidioschyzon merolae Debaryomyces hansenii Drosophila melanogaster Encephalitozoon cuniculi Entamoeba histolytica Plants Animal s Protists Fungi http://www.ncbi.nlm.nih.gov/genomes/leuks.cgi
Genomes Complete • Eukaryotes--23 complete, 329 in assembly, in progress 389 • Human, mouse, rat, zebrafish, • Homo sapiens neanderthalensis • Drosophila, Anopheles, Caenorhabditis • Arabadopsis,oat, corn, barley, rice, tomato • Saccharomyces, Schizosaccharomyces,Magnaportha, Cryptococcus, Candida… • Encephalitozoon cuniculi, Guillardia theta • Toxoplasma, Plasmodium • And many more…
Eubacteria and Archaea genomes • 608 Bacteria and 48 Archaea completed • Comprehensive Microbial Resource • http://pathema.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi • Joint Genome Institute • http://www.jgi.doe.gov/genome-projects/ • 2065 genome projects underway or completed! • NCBI Genomes
Genome Centers • Joint Genome Institute (DOE) • Whitehead Institute (MIT) • TIGR • Washington University (St. Louis) • Celera • Sanger Institute (the other UK) • RIKEN (Japan) • Beijing Genomics Institute (China) • Max Planck (Germany) …
Where do you find Genomic data? • NCBI • Entrez (by clone, by Refseq) • Genome (view and search map) • Genome center sites • Organism genome project sites • Annotations projects • UCSC Genome Browser, • Ensembl Genome Browser
Arabidopsis http://mips.helmholtz-muenchen.de/plant/athal/index.jsp
C. elegans (nematode) http://wormbase.org