Download

1 / 18
210 likes | 586 Views
Hemoglobin ( Hb ) Hb is found in RBCs its main function is to transport O 2 to tissues. Structure: 2 parts : heme + globin Globin: four globin chains (2 α and 2 β ).

E N D
Hemoglobin (Hb) Hb is found in RBCs its main function is to transport O2 to tissues. Structure: 2 parts : heme + globin Globin: four globin chains (2 α and 2β). Heme: Porphyrin ring with central iron. Iron is the site of attachment with O2.Iron must be in ferrous state to bind oxygen and form oxy Hb. Ferric state can’t carry oxygen and it form methemoglobin (MetHb) There are 4 heme groups each attached to one globin chain. So one Hb molecule can carry up to 4 O2 molecules and in this case called oxyHb.
Types of Hb: Hb A or HbA1 (Adult Hb): is the normal Hb in adults represents about 97% of total Hb. it is composed of 2 α and 2 β chains. HbA2: minor adult Hb, comprised 3% of normal adult Hb. Composed of 2 α and 2 δ chains HbF(fetal Hb): is the main Hb during fetal life and in newborns then disappear gradually where it is replaced by Hb A shortly after birth. It is composed of 2α and 2 γ chains. Hb F has greater affinity for O2 than HbA so ensure O2 transfer from maternal circulation to fetus RBCs through placenta. Note: The overall hemoglobin composition in a normal adult is approximately 97.5% HbA1, 2% HbA2 and 0.5% HbF.
Mutations in hemoglobin (hemoglobinopathies): 1- Sickle cell anemia (Hb S disease): It is a genetic disorder of blood caused by mutation in β-globin chain resulting in the formation of Hb S. The mutation occurs in 6th position of β-chain where glutamic acid is replaced by valine (non polar). Valine residues aggregate together by hydrophobic interactions leading to precipitation of Hb within RBCs. RBCs assume sickle-shaped leading to fragility of their walls and high rate of hemolysis.
Such sickled cells frequently block flow of blood in narrow capillaries and block blood supply to tissue (tissue anoxia) causing pain and cell death. Note: The life time of erythrocyte in sickle cell is less than 20 days, compared to 120 days for normal RBCs.
Patients may be : Heterozygotes (HbAS): There are two copies of the gene that synthesize βglobin chain. In Hb AS,mutation occurs only in one copy of the gene so patients have some normal Hb A. These patients have sickle cell trait with no clinical symptoms and can have normal life span. Or: Homozygotes (Hb SS): mutation occurs in both copies of gene that synthesize β-globin chain with apparent anemia and its symptoms
2- Hb C disease: Like HbS, Hb C is a mutant Hb in which glutamic acid in 6th position of β-chain is replaced by lysine. RBCs will be large oblong and hexagonal. The heterozygous form (HbAC) is asymptomatic. The homozygous form (Hb CC) causes anemia, tissue anoxia and severe pain.
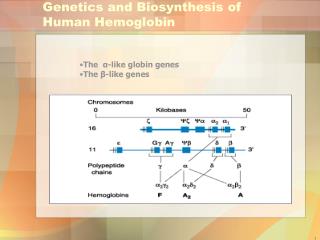
3- Thalassemia: A group of genetic diseases in which a defect occur in the rate of synthesis of one or more of Hb chains, but the chains are structurally normal. This due to defect or absence of one or more of • genes responsible for synthesis of α or β chains (see Fig.) leading to premature death of RBCs. • Types: • β -thalassemia: When synthesis of β chains is decreased or absent. • There are two copies of the gene responsible for synthesis of β chains. Individuals with β globin gene defects have either : • -β -thalassemia minor (β –thalassemia trait) : when the synthesis of only one β –globin gene is defective or absent. Those individuals make some β chains and usually not need specific treatment. • β -thalassemia major (Cooley anemia): if both genes are defective. • Babies will be severely anemic during the first or second year of life and so require regular blood transfusion. Bone marrow replacement is more safe treatment (why?).
Genes responsible for synthesis of Hb molecule (α and β globin chains)
α-thalassemia: in which synthesis of αglobin chain is defective or absent. There are four copies of gene responsible for synthesis of αglobin chains so patients may have: • i - Silent carrier of α-thalassemia with no symptoms: • if one copy of the genes is absent • ii- α-thalassemia trait: if two copies of genes are absent • iii- Hb H disease: if 3 copies of genes are absent, with mild to moderate anemia. The produced Hb will be β4 which is called Hb H. Oxygen delivery to tissues will be blocked because Hb H (β4 ) which bind O2 but does not deliver it to tissues. • iv- Hydropsfetalis: when all 4 copies of αglobin genes are absent. It causes fetal death (babies with this disorder usually die before or shortly after birth) because αglobin chains are required for synthesis of Hb F.
The inheritance of alpha thalassemia is complex because each parent potentially passes two of their four alpha globin genes to the offspring
Myoglobin: • It is a heme protein formed of one heme molecule attached to one globin chain (monomer). It can carry one molecule of oxygen that is bound to iron (ferrous state) • it is a monomer globular protein • its secondary structure contains high proportion (80%)of α helix. It contains 8 α-helix • It is found only in red skeletal muscles and cardiac muscle. It gives their tissues their characteristic red color. • Myoglobin serves as an intracellular storage site for oxygen. During periods of oxygen deprivation oxymyoglobin releases its bound oxygen which is then used for metabolic purposes within the cell. • So, It is used to store oxygen rather than transport it.
Clinical significance of myoglobin: • Myoglobin concentration is increased in blood in: • disease called myocardial infarction • and in case of muscle trauma and myopathies i.e. in any case related to damage of muscle tissues. • NB: Hb and myoglobin are hemoprotein that have ability to bind oxygen
Q: Certain amino acids are not part of primary structure of proteins but are synthesized after translation by certain modification. In collagen, which amino acid is not a part of the pimary structure of the protein? a) proline b) glycine c) hydroxyproline d) lysine
Question II: Give the name of: 1- Three plasma proteins with transport function 2- A protein with storage function: 3- Globular protein with a quaternary structure: 4- Fibrous protein with a quaternary structure: