Download

1 / 28
280 likes | 294 Views
Protein docking is vital for understanding molecular recognition in cells. Learn about computational tools, therodynamics, energy components, and post-processing for predicting near-native structures.

E N D
An Integrated Approach to Protein-Protein Docking Zhiping Weng Department of Biomedical Engineering Bioinformatics Program Boston University
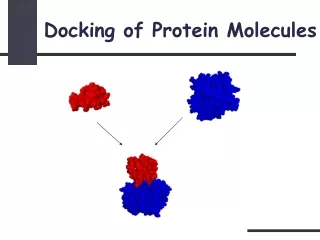
L L R R What is Protein Docking? Protein docking is the computational determination of protein complex structure from individual protein structures.
Motivation • Biological activity depends on the specific recognition of proteins. • Understand protein interaction networks in a cell • Yield insight to thermodynamics of molecular recognition • The experimental determination of protein-protein complex structures remains difficult.
Experimental Tools for Studying Protein-Protein Interactions • 3-D structures of protein-protein complexes: X-ray crystallography & NMR • Binding affinity between two proteins: SPR, titration assays • Mutagenesis and its affect on binding • Yeast 2-hybrid system • Protein Chips?
Computational Tools for Studying Protein-Protein Interactions • Protein docking • Binding affinity calculation • Analysis of site-specific mutation experiments • Protein design • The kinetics of protein-protein interactions
water L L L L L L L R R R L R R R Protein-Protein Interaction Thermodynamics
L L L L R R L R R R The Lowest Binding Free Energy DG water
Two kinds of docking problems • Bound docking The complex structure is known. The receptor and the ligand in the complex are pulled apart and reassembled. • Unbound docking Individually determined protein structures are used.
Challenges • Large search space • Imperfect understanding of thermodynamics • Protein flexibility • Heterogeneities in protein interactions
Divide and Conquer • Initial stage of unbound docking • Assume minimum binding site information • Try to predict as many near-native structures (hits) as possible in the top 1000, for as many complexes as possible • Post-processing • Re-rank the 1000 structures in order to pick out near-native structures
van der Waals energy; Shape complementarity Desolvation energy; Hydrophobicity Electrostatic interaction energy Translational, rotational and vibrational free energy changes An Effective Binding Free Energy Function Number of atoms of type i Desolvation energy for an atom of type i
R FFT Correlation IFFT FFT Y X L Surface Interior Binding Site Fast Fourier Transform Increase the speed by 107
Evaluate Performance • Gold Standard: Crystal structure of the complex • A near-native structure (hit):RMSD of Ca after superposition < 2.5 Å • Success rate: Given some number of predictions, percentage of complexes with at least one hit
Docking Benchmark 55 non-redundant complexes http://zlab.bu.edu/~rong/dock/
Target 2: Antibody/VP6 Red: Crystal Structure Blue: Prediction 50/52; 1st
Target 7:T Cell Receptor / Toxin Red: Crystal Structure Blue: Prediction 31/37, 1st
Target 3:Antibody/Hemagglutinin Red: Crystal Structure Blue: Prediction 37/62, 3rd
Target 6:Camelide Antibody/a amylase Red: Crystal Structure Blue: Prediction 18/65
Target 1:Hpr/HPrK Red: Crystal Structure Blue: Prediction 5/52
Summary • Conformational change tolerant target functions are needed for unbound docking • We need to balance shape complementarity, desolvation, electrostatics components • If we submit 10 predictions, we have a 60% success rate.
Future Work • An automatic protein-protein docking server • Large scale comparison of all docking algorithms on the benchmark • Post processing with binding site information, conformation space search, clustering and detailed free energy calculation • Make predictions!