Download

1 / 40
450 likes | 1k Views
Protein Analysis Workshop 2010. Secondary Structure Prediction. Bioinformatics group Institute of Biotechnology University of helsinki. Earlier version: Hung Ta Current: Petri Törönen. Overview. Hierarchy of protein structure. Introduction to structure prediction: Different approaches.

E N D
ProteinAnalysisWorkshop 2010 Secondary Structure Prediction Bioinformatics group Institute of Biotechnology University of helsinki Earlier version: Hung Ta Current: Petri Törönen
Overview • Hierarchy of protein structure. • Introduction to structure prediction: • Different approaches. • Prediction of 1D strings of structural elements. • Server/soft review: • COILS, MPEx, … • The PredictProtein metaserver.
Proteins • Proteins play a crucial role in virtually all biological processes with a broad range of functions. • Protein structure leads to protein function.
Primary Structure: a Linear Arrangement of Amino Acids • An amino acid has several structural components: a central carbon atom (Ca), an amino group (NH2), a carboxyl group (COOH), a hydrogen atom (H), a side chain (R). There are 20 amino acids • The peptide bond is formed as the cacboxyl group of an aa bind to the amino group of the adjacent aa. • The primary structure of a protein is simply the linear arrangement, or sequence, of the amino acid residues that compose it
major internal supportive elements, 60 percent of the polypeptide chain Secondary Structure: Core Elements of Protein Architecture • resulted from the folding of localized parts of a polypeptide chain. • α-helix • β-sheet • Coils, turns,
α-Helix • Hydrogen-bonded • 3.6 residues per turn • Axial dipole moment • Side chains point outward • Average length is 10 amino acids (3 turns). • Typically, rich of Analine, Glutamine, Leucine, Methione; and poor of Proline, Glycine, Tyrosine and Serine.
Ribbon diagram parallel anti-parallel β-Sheet • Formed due to hydrogen bonds between β-strands which are short polypeptide segments (5-8 residues). • Adjacentβ-strands run in the same directions -> parallel sheet. • Adjacent β-strands run in the oposite directions -> anti-parallel sheet.
Turns, loops, coils… • A turn, composed of 3-4 residues, forms sharp bends that redirect the polypeptide backbone back toward the interior. • A loop is similar with turns but can form longer bends • Turns and loops help large proteins fold into compact structures. • A random coil is a class of conformations that indicate an absence of regular secondary structure. Turn
Tertiary Structure: Overall Folding of Polypeptide Chain. • stabilized by hydrophobic interactions between the nonpolar side chains, hydrogen bonds between polar side chains, and peptide bonds
Quaternary Structure: Arrangement of Multiple Folded Protein Molecules. DNA polymerase Hemoglobin
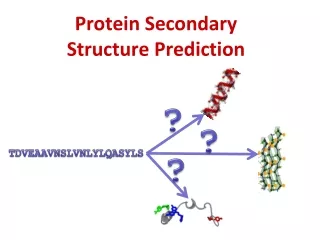
Structure Prediction GPSRYIVDL… ? • High importance in medicine (for example, in drug design) and biotechnology (for example, in the design of novel enzymes)
Structure Prediction • Why: experimentalmethods, X-ray crystallography or NMR spectroscopy, are very time-consuming and relatively expensive. • Structure is moreconservedthansequence. • Thiscanbeusefulwhenanalyzingunknownsequence. • Challenges: • Extremelylargenumber of possiblestructures. • the physical basis of protein structural stability is not fully understood.
Two options for structure prediction: • Ab initio (Start from scratch) • Homology modelling • If homology is found => use latter • If no homology => secondary structure can still be estimated
H: α-helix • E: β- strand • T: turn • C: coil aa Secondary Structure Prediction • Why: the first level of structural organization. • The tasks: Primary: MSEGEDDFPRKRTPWCFDDEHMC Secondary: CCHHHHHHCCCCEEEEEECCCCC
Secondary Structure Prediction Single residue statistical analysis (Chou-Fasman -1974): • For each amino acid type, assign its ‘propensity’ to be in a helix, β-sheet, or coil. • Based on 15 proteins of known conformation, 2473 total amino acids. • Limited accuracy: ~55-60% on average. • Eg: Chou-Fasman (1974), not used any more
Secondary Structure Prediction Segment-based statistics: • Look for correlations (within 11-21 aa windows). • Many algorithms have been tried. • Most performant: Neural Networks: • Input: a number of protein sequences with their known secondary structure. • Output: a trained network that predicts secondary structure elements for given query sequences. • Accuracy < 70%.
POPULAR SERVERS FOR DEALING WITH SECONDARY STRUCTURES • Coiled-coils • Transmembrane helices • Secondary structure • Metaservers
Prediction of coiled-coils Coiled-coils are generally solvent exposed multi-stranded helix structures: two-stranded Helix periodicity and solvent exposure impose special pattern of heptad repeat: Helical diagram of 2 interacting helices: … abcdefg … • hydrophobic residues • hydrophilic residues (From Wikipedia Leucine zipper article)
The COILS server at EMBnet • Compares a sequence to a database of known, parallel two-stranded coiled-coils, and derives a similarity score. • By comparing this score to the distribution of scores in globular and coiled-coil proteins, the program then calculates the probability that the sequence will adopt a coiled-coil conformation. • Options: • scoring matrices, • window size (score may vary), • weighting options.
COILS Limitations • The program works well for parallel two-stranded structures that are solvent-exposed but runs progressively into problems with the addition of more helices, their antiparallel orientation and their decreasing length. • The program fails entirely on buried structures.
COILS Demo Let us submit the sequence >1jch_A VAAPVAFGFPALSTPGAGGLAVSISAGALSAAIADIMAALKGPFKFGLWGVALYGVLPSQ IAKDDPNMMSKIVTSLPADDITESPVSSLPLDKATVNVNVRVVDDVKDERQNISVVSGVP MSVPVVDAKPTERPGVFTASIPGAPVLNISVNNSTPAVQTLSPGVTNNTDKDVRPAFGTQ GGNTRDAVIRFPKDSGHNAVYVSVSDVLSPDQVKQRQDEENRRQQEWDATHPVEAAERNY ERARAELNQANEDVARNQERQAKAVQVYNSRKSELDAANKTLADAIAEIKQFNRFAHDPM AGGHRMWQMAGLKAQRAQTDVNNKQAAFDAAAKEKSDADAALSSAMESRKKKEDKKRSAE NNLNDEKNKPRKGFKDYGHDYHPAPKTENIKGLGDLKPGIPKTPKQNGGGKRKRWTGDKG RKIYEWDSQHGELEGYRASDGQHLGSFDPKTGNQLKGPDPKRNIKKYL to the COILS server at EMBnet: http://www.ch.embnet.org/software/COILS_form.html
Correct answer: http://www.rcsb.org/pdb/explore/explore.do?structureId=1JCH
Transmembrane Region Prediction Transmembrane regions: • Usually contain residues with hydrophobic side chains (surface must be hydrophobic). • Usually ~20 residues long, can be up to 30 if not perpendicular through membrane. Methods: • Hydropathy plots (historical, better methods now available) • Threading (TMpred, MEMSAT), • Hidden Markov Model (TMHMM), • Neural Network (PHDhtm).
Hydropathy Plots (Kyte-Doolittle) • The hydropathy index of an amino acid is a number representing the hydrophobic or hydrophilic properties of its side-chain • compute an average hydropathy value for each position in the query sequence, • window length of 19 usually chosen for membrane-spanning region prediction.
Hydropathy Plot Servers Let us submit the sequence >sp|P06010|RCEM_RHOVI Reaction center protein M chain (Photosynthetic reaction center M subunit) - Rhodopseudomonas viridis. ADYQTIYTQIQARGPHITVSGEWGDNDRVGKPFYSYWLGKIGDAQIGPIYLGASGIAAFAFGSTAILIILFNMAAEVHFDPLQFFRQFFWLGLYPPKAQYGMGIPPLHDGGWWLMAGLFMTLSLGSWWIRVYSRARALGLGTHIAWNFAAAIFFVLCIGCIHPTLVGSWSEGVPFGIWPHIDWLTAFSIRYGNFYYCPWHGFSIGFAYGCGLLFAAHGATILAVARFGGDREIEQITDRGTAVERAALFWRWTIGFNATIESVHRWGWFFSLMVMVSASVGILLTGTFVDNWYLWCVKHG AAPDYPAYLPATPDPASLPGAPK to • Membrane Explorer (also as standalone MPEx), • Grease (http://fasta.bioch.virginia.edu/fasta_www2/fasta_www.cgi?rm=misc1) • Remove the FASTA header, if seq reading is not working.
Hydropathy Plot • The larger the number is, the more hydrophobic the amino acid • Correct answer (http://pir.uniprot.org/uniprot/P06010)
TM Pred Method summary: • Scans a candidate sequence for matches to a sequence scoring matrix, obtained by aligning the sequences of all transmembrane alpha-helical regions that are known from structures. • These sequences are collected in a database called TMBase. Remark: Authors do not suggest this method for genomic sequences. Automatic methods recommended, eg, TMHMM, PHDhtm.
TM Pred Server Let us submit RCEM_RHOVI again >sp|P06010|RCEM_RHOVI Reaction center protein M chain (Photosynthetic reaction center M subunit) - Rhodopseudomonas viridis. ADYQTIYTQIQARGPHITVSGEWGDNDRVGKPFYSYWLGKIGDAQIGPIYLGASGIAAFAFGSTAILIILFNMAAEVHFDPLQFFRQFFWLGLYPPKAQYGMGIPPLHDGGWWLMAGLFMTLSLGSWWIRVYSRARALGLGTHIAWNFAAAIFFVLCIGCIHPTLVGSWSEGVPFGIWPHIDWLTAFSIRYGNFYYCPWHGFSIGFAYGCGLLFAAHGATILAVARFGGDREIEQITDRGTAVERAALFWRWTIGFNATIESVHRWGWFFSLMVMVSASVGILLTGTFVDNWYLWCVKHG AAPDYPAYLPATPDPASLPGAPK to the TMPred server at EMBnet: http://www.ch.embnet.org/software/TMPRED_form.html
Meta-Servers • allows you to obtain many informations based on your sequence including structure predictions, motif or domain search… The predictions are based on several methods. • PredictProtein: http://predictprotein.org A server which
The PredictProtein meta-server • For sequence analysis, structure and function prediction. When you submit any protein sequence PredictProtein retrieves similar sequences in the database and predicts aspects of protein structure and function • SEG: finds low complexity regions. • ProSite: database of functional motifs, ie, biologically relevant short patterns • ProDom: a comprehensive set of protein domain families automatically generated from the SWISS-PROT and TrEMBL sequence databases. • PROFsec (PHDsec): secondary structure, • PROFacc (PHDacc): solvent accessibility, • PHDhtm: transmembrane helices. • Sequence database is scanned for similar sequences (Blast, Psi-Blast). • Multiple sequence alignment profiles are generated by weighted dynamic programming (MaxHom).
PredictProtein Demo Let´s submit again >uniprot|P00772|ELA1_PIG Elastase-1 precursor MLRLLVVASLVLYGHSTQDFPETNARVVGGTEAQRNSWPSQISLQYRSGSSWAHTCGGTL IRQNWVMTAAHCVDRELTFRVVVGEHNLNQNDGTEQYVGVQKIVVHPYWNTDDVAAGYDI ALLRLAQSVTLNSYVQLGVLPRAGTILANNSPCYITGWGLTRTNGQLAQTLQQAYLPTVD YAICSSSSYWGSTVKNSMVCAGGDGVRSGCQGDSGGPLHCLVNGQYAVHGVTSFVSRLGC NVTRKPTVFTRVSAYISWINNVIASN to http://predictprotein.org/ For a list of mirror sites: http://predictprotein.org/newwebsite/doc/mirrors.html
Low-complexity regions Marked by ’X’
References • Documentation: • COILS:http://www.ch.embnet.org/software/coils/COILS_doc.html • TMPred:http://www.ch.embnet.org/software/tmbase/TMBASE_doc.html • MPEx:http://blanco.biomol.uci.edu/mpex/MPEXdoc.html • Articles: • B. Rost: Evolution teaches neural networks. In Scientific applications of neural nets. Ed. J.W.Clark, T.Lindenau, M.L. Ristig, 207-223 (1999). • D.T Jones: Protein Secondary Structure Prediction Based on Position-specific Scoring Matrices. J.Mol.Biol. 292, 195-202 (1999). • B. Rost: Prediction in 1D: Secondary Structure, Membrane Helices, and Accessibility. In Structural Bioinformatics (reference below). • Books: • P.E. Bourne, H. Weissig: Structural Bioinformatics. Wiley-Liss, 2003. • A. Tramontano: Protein Structure Prediction. Wiley-VCH, 2006.