Download

1 / 88
910 likes | 1.14k Views
U n i v e r s i t a t P o m p e u F a b r a. Baldomero Oliva Miguel. Comparative Modelling Threading. EVOLUTION. Diferences (proteins). Genetic information (DNA). DNA secuence 3D. U.S. Genome Project ( 1990 ). Identify human genes.

E N D
U n i v e r s i t a t P o m p e u F a b r a Baldomero Oliva Miguel Comparative Modelling Threading
Diferences (proteins) Genetic information (DNA)
U.S. Genome Project ( 1990 ) Identify human genes Store the information in Data Banks Development of tools for analisis 1998 DOE The Sanger Centre Venter, Patrinos & Collins NIH NHGRI
Increase of sequences 10500000 GenBank (DNA) 378152 TrEMBL 92211 Swiss Prot
sec. 3D 3D known (20%) Unknown function (20%) Homologs (30%) NO homology (20%) Remotes (10%)
Knowing the structure helps to: • TO KNOWthe function: annotation • IMPROVE the function: genetic engineering • INHIBIT the function: drugs
Structure Factory express & purify cristallize X-ray analises structure
NO information 3D 3D 3D Enough information 3D Objetive Not accurate
How does modelling help? Structural Genomics X-ray
Study of known structures Assigning structure to sequences with unknown structure
Caracterize the conformation sequence quaternary secondary Ligand docking Met Val Leu Tyr Asp domain Ser Thr • • • Tertiary super-secondary
Mioglobin Carboxypeptidase Triose Fosfate Isomerase Ribonuclease Glucanase
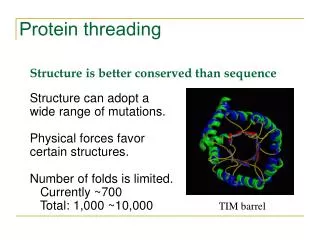
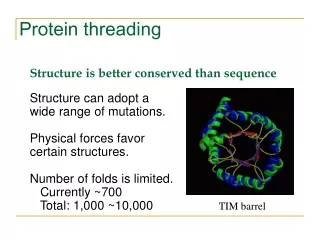
The number of different protein folds is limited: Known Folds New Folds
The number of different protein folds is limited: [ last update: Oct 2001 ]
Nº sequences > Nº folds Structura 3ª Classification
Protein Structure Classification CATH - Protein Structure Classification [ http://www.biochem.ucl.ac.uk/bsm/cath_new/ ] • UCL, Janet Thornton & Christine Orengo • Class (C), Architecture(A), Topology(T), Homologous superfamily (H) SCOP - Structural Classification of Proteins • MRC Cambridge (UK), Alexey Murzin, Brenner S. E., Hubbard T., Chothia C. • created by manual inspection • comprehensive description of the structural and evolutionary relationships [ http://scop.mrc-lmb.cam.ac.uk/scop/ ]
Class(C)derived from secondary structure content is assigned automatically • Architecture(A)describes the gross orientation of secondary structures, independent of connectivity. • Topology(T) clusters structures according to their topological connections and numbers of secondary structures • Homologous superfamily (H)
Homolog Cholera toxin 80% Id Remote TSS toxin 8.8% Id Analog Aminoacyl tRNA synthetase 4.4% Id Enterotoxin SCOP • Family • Superfamily • Fold • Class
MNIFEMLRID EGLRLKIYKD TEGYYTIGIG HLLTKSPSLN AAKSELDKAI GRNCNGVITK DEAEKLFNQD VDAAVRGILR NAKLKPVYDS LDAVRRCALI NMVFQMGETG VAGFTNSLRM LQQKRWDEAA VNLAKSRWYN QTPNRAKRVI TTFRTGTWDA YKNL Can we predict protein structures ?
Homology modeling Idea:Extrapolation of the structure for a new (target) sequence from the known 3D-structures of related family members (templates).
Nº sequences > Nº folds Alignments Step 1 Selection of templates Step 2 Alignment with templates
1 0 0 i d e n t i t y 8 0 Sequence identity implies structural similarity 6 0 identity/similarity Percentage sequence 4 0 Don’t know region ..... 2 0 0 (B.Rost, Columbia, NewYork) 0 5 0 1 0 0 1 5 0 2 0 0 2 5 0 Number of residues aligned Sequence similarity implies structural similarity? .
Nº sequences > Nº folds 2D Structure Hidropaticity Plot 2D prediction
Nº sequences > Nº folds 2D Structure Hidropaticity Plot 2D prediction Structure 3D Alignments Classification - Homologs - Remotes - Analogs
Comparative Modelling (homology) • Fold is more conserved than sequence. • Secondary structure is the most conserved structure • Loops have the higher variability in structure.
Nº sequences > Nº folds 2D Structure Hidropaticity Plot 2D prediction Structure 3D Alignments Classification - Homologs - Remotes - Analogs Comparative modelling - Composer - Modeler SCR & VR SCR VR - Ring Closure - DB Search - Classification
Nº sequences > Nº folds 2D Structure Hidropaticity Plot 2D prediction Structure 3D Alignments Classification - Homologs - Remotes - Analogs Comparative modelling - Composer - Modeler SCR & VR SCR MODEL VR - Ring Closure - DB Search - Classification Step 3 Model Building
Rigid-body Assembly Spatial Constrains Fragment Extraction Satisfaction of Spatial Constrains Loop Construction Optimisation Methods
Rigid Body assembly a) Build conserved core framework • averaging core template backbone atoms (weighted by local sequence similarity with the target sequence) • Leave non-conserved regions (loops) for later ….
Rigid Body assembly b) Loop modeling • use the “spare part” algorithm to find compatible fragments in a Loop-Database • “ab-initio” rebuilding of loops (Monte Carlo, molecular dynamics, genetic algorithms, etc.)
D/N 100% D/N 100% D 100% aa{baalal}bb Xh{DXDpDG}Xh EF-Hand Calcium binding aa{baalal}bb Xh{DXDpDG}Xh
P-loop GTP binding P-loop GTP binding Conformation bb{eppgag}aa Motif sequence hh{GhXXpG}Kp Conformation bb{eppgag}aa Motif sequence hh{GhXXpG}Kp
bb{eab}aa hh{GhG}hX NAD(P)/FAD binding bb{eab}aa hh{GhG}hX
Ring closure search Distance restraints
Building CA Superposition
Building CA Superposition
Q M T S A F G T A E Modeling by Satisfaction of Spatial restraints A. Sali & T. Blundel (1993) JMB 234, 779 - 815
Modeling by Satisfaction of Spatial restraints • Feature properties can be associated with • a protein (e.g. X-ray resolution) • residues (e.g. solvent accessibility) • pairs of residues (e.g. Ca - Ca distance) • other features (e.g. main chain classes) How can we derive modeling restraints from this data? A restraint is defined as probability density function (pdf) p(x): with
Modeling by Satisfaction of Spatial restraints combine basis pdfs to molecular probability density functions
Modeling by Satisfaction of Spatial restraints • Satisfaction of spatial restraints • Find the protein model with the highest probability • Variable target function: • Start with e.g. a random conformation model and use only local restraints • minimize some steps using a conjugate gradient optimization • repeat with introducing more and more long range restraints until all restraints are used
Threading Idea:Find the optimal structure for a new (target) sequence in the set of known 3D-structures (templates) by threading the target sequence.
Nº sequences > Nº folds Structure 3D Classification - Homologs - Remotes - Analogs Step 1 Selection of templates FOLD ASSIGNMENT
Nº sequences > Nº folds Structure 3D Classification - Homologs - Remotes - Analogs Pseudo Potencial
Pseudo-potencial Principles Boltzman law Potential of Mean Force: Applied on proteins Use frequencies instead of probabilities. They are taken from non-homologous proteins on the PDB.
? Fold recognition / Threading Principle: Find a compatible fold for a given sequence .... >Protein XY MSTLYEKLGGTTAVDLAVDKFYERVLQDDRIKHFFADVDMAKQRAHQKAFLTYAFGGTDKYDGRYMREAHKELVENHGLNGEHFDAVAEDLLATLKEMGVPEDLIAEVAAVAGAPAHKRDVLNQ • Using ... • 1D – 3D profile matching, • secondary structure predictions, • position specific scoring matrices (PSSM), • mean force potentials, • keyword statistics, • ....