Download
1 / 16
320 likes | 942 Views
Cystic Fibrosis. A Presentation Constructed by Stacy Salerno. Clinical Features. Cystic fibrosis is a heterogeneous recessive genetic disorder with features that reflect mutations in the cystic fibrosis transmembrane conductance regulator ( CFTR ) gene.

E N D
Cystic Fibrosis A Presentation Constructed by Stacy Salerno
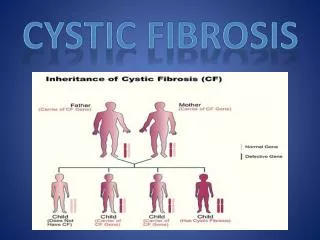
Clinical Features • Cystic fibrosis is a heterogeneous recessive genetic disorder with features that reflect mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. • Classic cystic fibrosis is characterized by chronic bacterial infection of the airways and sinuses, fat maldigestion due to pancreatic exocrine insufficiency, infertility in males due to obstructive azoospermia, and elevated concentrations of chloride in sweat. • Patients with nonclassic cystic fibrosis have at least one copy of a mutant gene that confers partial function of the CFTR protein, and such patients usually have no overt signs of maldigestion because some pancreatic exocrine function is preserved.
Genotype and Phenotype • Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene which encodes a protein expressed in the apical membrane of exocrine epithelial cells. • This genotypic variation provides a rationale for phenotypic effects of the specific mutations. The extent to which various CFTR alleles contribute to clinical variation in CF is evaluated by genotype-phenotype studies. • The poor correlation between CFTR genotype and severity of lung disease strongly suggests an influence of environmental and secondary genetic factors (CF modifiers). • Several candidate genes related to innate and adaptive immune response have been implicated as pulmonary CF modifiers. In addition, the presence of a genetic CF modifier for meconium ileus has been demonstrated on human chromosome 19q13.2. • The phenotypic spectrum associated with mutations in the CFTR gene extends beyond the classically defined CF. Besides patients with atypical CF, there are large numbers of so-called monosymptomatic diseases such as various forms of obstructive azoospermia, idiopathic pancreatitis or disseminated bronchiectasis associated with CFTR mutations uncharacteristic for CF.
Molecular Genetics and Gene Function • Locus: 7q31.2 - The CFTR gene is found in region q31.2 on the long (q) arm of human chromosome 7. • Gene Structure: The normal allelic variant for this gene is about 250,000 bp long and contains 27 exons. • mRNA: The intron-free mRNA transcript for the CFTR gene is 6129 bp long. • Coding Sequence (CDS): 4443 bp within the mRNA code for the amino acid sequence of the gene's protein product. • Protein Size: The CFTR protein is 1480 amino acids long and has a molecular weight of 168,173 Da. • Protein Function: The normal CFTR protein product is a chloride channel protein found in membranes of cells that line passageways of the lungs, liver, pancreas, intestines, reproductive tract, and skin. CFTR is also involved in the regulation of other transport pathways. • Associated Disorders: Defective versions of this protein, caused by CFTR gene mutations, can lead to the development of cystic fibrosis (CF) and congenital bilateral aplasia of the vas deferens (CBAVD).
Protein Function and Biochemistry • CFTR controls chloride ion movement in and out of the cell.
Protein Structure and Function • CFTR transports chloride ions (Cl-) ions across the membranes of cells in the lungs, liver, pancreas, digestive tract, reproductive tract, and skin. • CFTR is made up of five domains: • two membrane-spanning domains (MSD1 and MSD2) that form the chloride ion channel • two nucleotide-binding domains (NBD1 and NBD2) that bind and hydrolyze ATP (adenosine triphosphate) • and a regulatory (R) domain. • Delta F508, the most common CF-causing mutation, occurs in the DNA sequence that codes for the first nucleotide-binding domain (NBD1).
Changes in Protein structure • CFTR functions principally as a cAMP-induced chloride channel and appears capable of regulating other ion channels. • Besides the most common mutation, ΔF508, accounting for about 70% of CF chromosomes worldwide, more than 850 mutant alleles have been reported to the CF Genetic Analysis Consortium. • These mutations affect CFTR through a variety of molecular mechanisms which can produce little or no functional CFTR at the apical membrane.
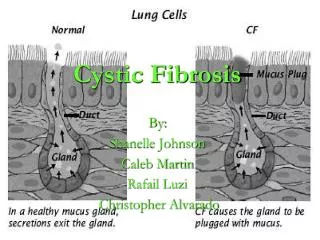
3D Image of Protein • When a CFTR protein with the delta F508 mutation reaches the ER, the quality-control mechanism of this cellular component recognizes that the protein is folded incorrectly and marks the defective protein for degradation. As a result, delta F508 never reaches the cell membrane. • People who are homozygous for delta F508 mutation tend to have the most severe symptoms of cystic fibrosis due to critical loss of chloride ion transport. • This upsets the sodium and chloride ion balance needed to maintain the normal, thin mucus layer that is easily removed by cilia lining the lungs and other organs. The sodium and chloride ion imbalance creates a thick, sticky mucus layer that cannot be removed by cilia and traps bacteria, resulting in chronic infections.
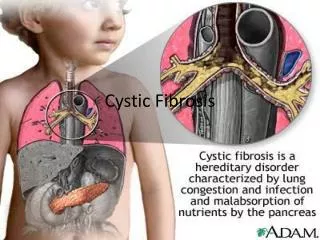
Presentation of Disease Mucous in the airways cannot be easily cleared from the lungs.
Presentation of Disease Colon Pancreas Sticky mucus secretion Ducts are filled with sticky mucus. Scaring of tissue.
Treatment • The only way to cure CF would be to use gene therapy to replace the defective gene or to give the patient the normal form of the protein before symptoms cause permanent damage. • The major goal in treating CF is to clear the abnormal and excess secretions and control infections in the lungs, and to prevent obstruction in the intestines. • For patients with advanced stages of the disease, a lung transplant operation may be necessary. • Although treating the symptoms does not cure the disease, it can greatly improve the quality of life for most patients and has, over the years, increased the average life span of CF patients to 30 years. Gastrointestinal Treatment • Modified dietDue to pancreatic disorders, children with CF require a modified diet, including vitamin supplements (vitamins A, D, E, and K) and pancreatic enzymes. Maintaining adequate nutrition is essential. The diet calls for a high-caloric content (twice what is considered normal for the child's age), which is typically low in fat and high in protein. Patients or their caregivers should consult with their health care providers to determine the most appropriate diet.
Gene Therapy • Gene therapy is the use of normal DNA to "correct" for the damaged genes that cause disease. • In the case of CF, gene therapy involves inhaling a spray that delivers normal DNA to the lungs. • The goal is to replace the defective CF gene in the lungs to cure CF or slow the progression of the disease.
References • http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Search&db=books&doptcmdl=GenBookHL&term=molecular+genetics+of+cystic+fibrosis+AND+gene%5Bbook%5D+AND+408389%5Buid%5D&rid=gene.chapter.cf#cf._Cystic_Fibrosis_2 • http://www.ionchannels.org/showabstract.php?pmid=9922375 • http://www.ornl.gov/sci/techresources/Human_Genome/posters/chromosome/cftr.shtml • UVM Pathology 101 Lecture on Childhood Disease • www.google.image.com • Noone PG, Pue CA, Zhou Z, Friedman KJ, Wakeling EL, Ganeshananthan M, Simon RH, Silverman LM, Knowles MR. Lung disease associated with the IVS8 5T allele of the CFTR gene. Am J Respir Crit Care Med. 2000. 162:1919-24. (PubMed) • Wang Z, Milunsky J, Yamin M, Maher T, Oates R, Milunsky A. Analysis by mass spectrometry of 100 cystic fibrosis gene mutations in 92 patients with congenital bilateral absence of the vas deferens. Hum Reprod. 2002. 17:2066-72. (PubMed) • Kiesewetter S, Macek M Jr, Davis C, Curristin SM, Chu CS, Graham C, Shrimpton AE, Cashman SM, Tsui LC, Mickle J, et al. A mutation in CFTR produces different phenotypes depending on chromosomal background. Nat Genet. 1993. 5:274-8. (PubMed)