Download

1 / 43
670 likes | 2.46k Views
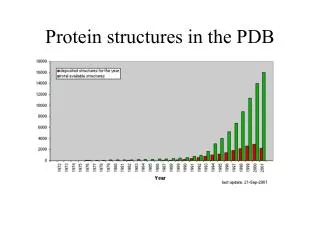
The Protein Data Bank (PDB). PDB is the principal repository for protein structures Established in 1971 Accessed at http://www.rcsb.org/pdb or simply http://www.pdb.org Currently contains over 32,000 structure entities. Updated 9/05. Page 287. PDB content growth (www.pdb.org).

E N D
The Protein Data Bank (PDB) • PDB is the principal repository for protein structures • Established in 1971 • Accessed at http://www.rcsb.org/pdb or simply • http://www.pdb.org • Currently contains over 32,000 structure entities Updated 9/05 Page 287
PDB content growth (www.pdb.org) structures year Fig. 9.6 Page 281
PDB holdings (September, 2005) 29,876 proteins, peptides 1,338 protein/nucl. complexes 1,500 nucleic acids 13carbohydrates 32,727 total Table 9-2 Page 281
gateways to access PDB files Swiss-Prot, NCBI, EMBL Protein Data Bank CATH, Dali, SCOP, FSSP databases that interpret PDB files Fig. 9.10 Page 285
Access to PDB through NCBI • You can access PDB data at the NCBI several ways. • Go to the Structure site, from the NCBI homepage • Use Entrez • Perform a BLAST search, restricting the output • to the PDB database Page 289
Access to PDB through NCBI Molecular Modeling DataBase (MMDB) Cn3D (“see in 3D” or three dimensions): structure visualization software Vector Alignment Search Tool (VAST): view multiple structures Page 291
Fig. 9.15 Page 290
Fig. 9.15 Page 290
Fig. 9.16 Page 291
Fig. 9.16 Page 291
Fig. 9.16 Page 291
Fig. 9.16 Page 291
Fig. 9.16 Page 291
Fig. 9.17 Page 292
Access to structure data at NCBI: VAST Vector Alignment Search Tool (VAST) offers a variety of data on protein structures, including -- PDB identifiers -- root-mean-square deviation (RMSD) values to describe structural similarities -- NRES: the number of equivalent pairs of alpha carbon atoms superimposed -- percent identity Page 294
Many databases explore protein structures SCOP CATH Dali Domain Dictionary FSSP Page 293
Structural Classification of Proteins (SCOP) SCOP describes protein structures using a hierarchical classification scheme: Classes Folds Superfamilies (likely evolutionary relationship) Families Domains Individual PDB entries http://scop.mrc-lmb.cam.ac.uk/scop/ Page 293
Class, Architecture, Topology, and Homologous Superfamily (CATH) database CATH clusters proteins at four levels: C Class (a, b, a&b folds) A Architecture (shape of domain, e.g. jelly roll) T Topology (fold families; not necessarily homologous) H Homologous superfamily http://www.biochem.ucl.ac.uk/basm/cath_new Page 293
SCOP statistics (September, 2005) Class # folds # superfamilies # families All a 218 376 608 All b 144 290 560 a/b 136 222 629 a+b 279 409 717 … Total 945 1539 2845 Table 9-4 Page 298 a/b = parallel bsheets a+b = antiparallel b sheets
Fig. 9.23 Page 298
Fig. 9.24 Page 299
Fig. 9.25 Page 300
Fig. 9.25 Page 300
Fig. 9.26 Page 301
Fig. 9.27 Page 302
Fig. 9.28 Page 303
Dali Domain Dictionary Dali contains a numerical taxonomy of all known structures in PDB. Dali integrates additional data for entries within a domain class, such as secondary structure predictions and solvent accessibility. Page 302
Fig. 9.29 Page 303
Fig. 9.30 Page 304
Fig. 9.30 Page 304
Fig. 9.30 Page 304
Fold classification based on structure-structure alignment of proteins (FSSP) FSSP is based on a comprehensive comparison of PDB proteins (greater than 30 amino acids in length). Representative sets exclude sequence homologs sharing > 25% amino acid identity. The output includes a “fold tree.” http://www.ebi.ac.uk/dali/fssp Page 293
Fig. 9.31 Page 305
FSSP: fold tree Fig. 9.32 Page 306
Fig. 9.33 Page 307
Fig. 9.34 Page 307
Approaches to predicting protein structures There are about >20,000 structures in PDB, and about 1 million protein sequences in SwissProt/ TrEMBL. For most proteins, structural models derive from computational biology approaches, rather than experimental methods. The most reliable method of modeling and evaluating new structures is by comparison to previously known structures. This is comparative modeling. An alternative is ab initio modeling. Page 303-305
Approaches to predicting protein structures obtain sequence (target) fold assignment comparative modeling ab initio modeling Fig. 9.35 Page 308 build, assess model
Comparative modeling of protein structures [1] Perform fold assignment (e.g. BLAST, CATH, SCOP); identify structurally conserved regions [2] Align the target (unknown protein) with the template. This is performed for >30% amino acid identity over a sufficient length [3] Build a model [4] Evaluate the model Page 305
Errors in comparative modeling Errors may occur for many reasons [1] Errors in side-chain packing [2] Distortions within correctly aligned regions [3] Errors in regions of target that do not match template [4] Errors in sequence alignment [5] Use of incorrect templates Page 306
Comparative modeling In general, accuracy of structure prediction depends on the percent amino acid identity shared between target and template. For >50% identity, RMSD is often only 1 Å. Page 306
Fig. 9.36 Page 308 Baker and Sali (2000)
Comparative modeling Many web servers offer comparative modeling services. Examples are SWISS-MODEL (ExPASy) Predict Protein server (Columbia) WHAT IF (CMBI, Netherlands) Page 309