Download

1 / 54
600 likes | 871 Views
Yadollah Zahed Pasha (MD) Professor of Pediatrics & Neonatology, Non-Communicable Pediatric Diseases Research Center, Babol University of Medical Sciences, Babol, IR Iran,1396/6/22. Glucose-6-Phosphate Dehydrogenase Deficiency In Neonates. G6PD deficiency is;

E N D
Yadollah Zahed Pasha (MD) Professor of Pediatrics & Neonatology, Non-Communicable Pediatric Diseases Research Center, Babol University of Medical Sciences, Babol, IR Iran,1396/6/22 Glucose-6-Phosphate Dehydrogenase Deficiency In Neonates
G6PD deficiency is; one of the most common human enzymopathies And is estimated to affect 400 million people worldwide, with 11 million G6PD-deficient infants born each year. Nkhoma ET, Poole C, Vannappagari V, et al. The global prevalence of glucose-6phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis 2009;42:267–78. Bhutani VK, Zipursky A, Blencowe H, et al. Neonatal hyperbilirubinemia and Rhesus disease of the newborn: incidence and impairment estimates for 2010 at regional and global levels. Pediatr Res 2013;74:86–100.
G6PD deficiency is an allelic abnormality which is inherited in an X-linked recessive fashion. • Beutler E,Blood Rev.1996 • Cappllini MD,Lancet.2008
The G6PD gene is X-linked. located at the Xq28 region on the tip of the long armof the X- chromosome. Beutler E. Blood Rev. 1996
Discovered that G6PD Deficiency was a genetic disorder in 1953 by the late Dr. Beutler
Role of G6PD in protecting against oxidative stress. G6P, glucose-6-phosphate. The function G6PD in the red cell is to generate NADPH → reduced glutathione → protect the RBCs from the oxidative damage by H2O2
Prevalence Zahed Pasha.Y JBUMS. 1996
More than 400 variants of Enzyme have been identified.(Mutatiom) • Kotaka M,Actacrystallogr, 2005 • Capellani MD,Lancet.2005 • Mason PJ,Blood Rev.2007 • Zhao X,J Bioinform Comput Biol.2010
Frequency of G6PD deficiency Variation In The world Zahedpasha MD 2015
Adapted from Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008;371:64–74.
Classification of G6PD Variants * Wild type NNJ= Neonatal Jaundice AHA= Acute Hemolytic Anemia CNSHA= chronic non-spherocytic anemia
Clinical Features • G6PD deficiency is usually asymptomatic. • Neonatal jaundice. • Acute haemolytic anaemia in response to oxidant stress: drugs, fava beans or infections. • Chronic Nonspherocytic hemolytic Anemia (CNSHA)
Because G6PD is X-linked; Heterozygous Female infants can carry severe mutations and remain symptomless, Whereas hemizygous Male infants with class I mutations suffer from chronic nonspherocytic hemolytic anemia (CNSHA). Luzzatto L. Glucose 6-phosphate dehydrogenase deficiency: from genotype to phenotype. Haematologica 2006;91:1303–6. Corrons JV, Feliu E, Pujades MA, et al. Severe-glucose-6-phosphate dehydrogenase (G6PD) deficiency associated with chronic hemolytic anemia, granulocyte dysfunction, and increased susceptibility to infections: description of a new molecular variant (G6PD Barcelona). Blood 1982;59:428–34
Approximately 80% of newborns worldwide have some degree of hyperbilirubinemia and visible jaundice. • Severe cases of hyperbilirubinemia can progress to kernicterus and lead to permanent developmental disorders. • Several risk factors contribute to hyperbilirubinemia and kernicterus, including glucose-6-phosphate dehydrogenase (G6PD) deficiency Maisels MJ. Managing the jaundiced newborn: a persistent challenge. CMAJ 2015;187:335–43.
Major risk factors Predischarge bili in high-risk zone Jaundice in 1st 24 hrs Blood group incomp with + direct antiglobulin test, other known hemolytic disease (G6PD deficiency) Gestational age 35–36 wk Previous sibling received phototherapy Cephalohematoma or significant bruising Exclusive breastfeeding East Asian race Minor risk factors Bili in high intermed-risk zone Gestational age 37–38 wk Jaundice before discharge Previous sibling with jaundice Macrosomia infant with diabetic mother Maternal age ≥ 25 Male Decreased Risk Bili in low-risk zone ≥ 41 wks gestation Exclusive bottle feed Black race D/c from hospital > 72hrs Risk Factors for Severe Hyperbilirubinemia Zahedpasha MD 2015
GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY INCREASES THE RISK OF NEONATAL HYPERBILIRUBINEMIA AND KERNICTERUS Infants with G6PD deficiency are significantly more likely to develop hyperbilirubinemia. Kaplan M, Hammerman C. Glucose-6-phosphate dehydrogenase deficiency and severe neonatal hyperbilirubinemia: a complexity of interactions between genes and environment. Semin Fetal Neonatal Med 2010;15:148–56.
Hyperbilirubinemia & Clinical Outcomes: JAUNDICE ACUTE BILIRUBIN ENCEPHALOPATHY KERNICTERUS Deposits in skin and mucous membranes Unconjugated bilirubin deposits in the brain Permanent neuronal damage Zahedpasha MD 2015
Neonatal Jaundice is a major clinical problem globally, especially in the Asian and southeast Asian region. Ho NK, BAILLRERES Clin Haematol,1992
Severe Jaundice leading to Kernicterus or death in newborn is the most devastating consequence of G6PD deficiency. Kaplan M,ProcNaltAcadSci USA.1997
The incidence of severe hyperbilirubinemia is estimated to be 2 to 45 per 100,000 births, and of kernicterus to be 0.4 to 2.7 per 100,000 births in developed countries. Olusanya BO, Ogunlesi TA, Slusher TM. Why is kernicterus still a major cause of death and disability in low-income and middle-income countries? Arch Dis Child 2014;99:1117–21.
A recent meta-analysis of 5 studies including more than 20,000 subjects found that G6PD-deficient infants are almost 4 times more likely to develop hyperbilirubinemia and 3 times more likely to receive phototherapy compared with G6PD-normal infants. Liu H, Liu W, Tang X, et al. Association between G6PD deficiency and hyperbilirubinemia in neonates: a meta-analysis. Pediatr Hematol Oncol 2015;32:92–8.
Moreover, G6PD-deficient infants are more likely to develop kernicterus. In the United States, 20% of infants who develop kernicterus have G6PD deficiency, compared with an estimated 4% to 7% prevalence of G6PD deficiency in the average population. Watchko JF, Kaplan M, Stark AR, et al. Should we screen newborns for glucose-6phosphate dehydrogenase deficiency in the United States? J Perinatol 2013;33: 499–504. Johnson L, Bhutani VK, Karp K, et al. Clinical report from the pilot USA kernicterus registry (1992 to 2004). J Perinatol 2009;29(Suppl 1):S25–45.
Of infants with kernicterus, 15% of those with G6PD-deficiency died, compared with 1% mortality in the G6PD-normal infants. Johnson L, Bhutani VK, Karp K, et al. Clinical report from the pilot USA kernicterus registry (1992 to 2004). J Perinatol 2009;29(Suppl 1):S25–45.
In developing regions, access to phototherapy and exchange transfusions is often limited. Even in developed nations, despite these simple and accessible treatments, up to 6.6% of G6PD-deficient infants will develop kernicterus,and 12% to 50% of G6PD-deficient infants with kernicterus will die. .Furthermore, exchange transfusion leads to adverse events in 5% of infants, and death in 0.4% of infants. Olusanya BO, Slusher TM. Reducing the burden of severe neonatal jaundice in G6PD-deficient populations in low-income countries: are we doing enough? Int Health 2010;2:22–4. Weng Y-H, Chiu Y-W. Clinical characteristics of G6PD deficiency in infants with marked hyperbilirubinemia. J Pediatr Hematol Oncol 2010;32:11–4. Kaplan M, Bromiker R, Hammerman C. Severe neonatal hyperbilirubinemia and kernicterus: are these still problems in the third millennium? Neonatology 2011; 100:354–62. Kaplan M, Hammerman C. Glucose-6-phosphate dehydrogenase deficiency:a hidden risk for kernicterus. Semin Perinatol 2004;28:356–64. Muchowski KE. Evaluation and treatment of neonatal hyperbilirubinemia. Am Fam Physician 2014;89:873–8.
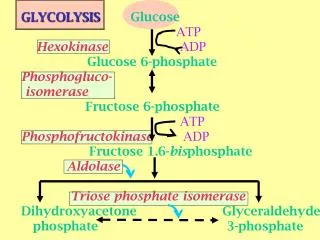
HOW DOES GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY CONTRIBUTE TO KERNICTERUS? • G6PD is the rate-limiting enzyme in the pentose phosphate pathway, catalyzing the oxidation of glucose-6-phosphate to 6-phospho-gluconate while reducing nicotinamide adenine dinucleotide phosphate (NADP) to NADPH. • NADPH then fuels the regeneration of reduced glutathione (GSH), which in turn neutralizes reactive oxygen species(ROS).
Bilirubin toxicity begins at the cell membranes, because bilirubin islipophilic and is highly concentrated in membrane compartments. Hansen T, Tommarello S, Allen J. Subcellular localization of bilirubin in rat brain after in vivo i.v. administration of [3H]bilirubin. Pediatr Res 2001;49:203–7
causing membrane permeability, leading to lipid peroxidation, and inhibiting the functions of membranebound proteins, such as ATPases. Similarly, bilirubin also targets mitochondrial membranes, leading to disruption of the electron transport chain and membranebound proteins, which, in turn, results in mitochondrial swelling, membrane permeability, depolarization, cytochrome c release, and cell death by apoptosis and Necrosis. Brito MA, Brites D, Butterfield DA. A link between hyperbilirubinemia, oxidative stress and injury to neocortical synaptosomes. Brain Res 2004;1026:33–43. Rodrigues CM, Sola´S, Castro RE, et al. Perturbation of membrane dynamics in nerve cells as an early event during bilirubin-induced apoptosis. J Lipid Res 2002;43:885–94. Keshavan P, Schwemberger SJ, Smith DL, et al. Unconjugated bilirubin induces apoptosis in colon cancer cells by triggering mitochondrial depolarization. IntJ Cancer 2004;112:433–45. Rodrigues CM, Sola´S, Silva R, et al. Bilirubin and amyloid-beta peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol Med 2000;6:936–46. Rodrigues CM, Sola´S, Brites D. Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatology 2002;35:1186–95. Oakes GH, Bend JR. Early steps in bilirubin-mediated apoptosis in murine hepatoma (Hepa 1c1c7) cells are characterized by aryl hydrocarbon receptorindependent oxidative stress and activation of the mitochondrial pathway. J BiochemMolToxicol 2005;19:244–55.
Contribution of G6PD deficiency to bilirubin-induced neurotoxicity. (A) Neonatal hyperbilirubinemia results from production of bilirubin (yellow hexagons) following hemolysis. In G6PD-normal infants, GSH levels are properly maintained and low levels of bilirubininduced oxidative stress are safely neutralized. (B) In G6PD-deficient infants, higher levels of hemolysis lead to higher concentration of bilirubin in the brain, which inhibits mitochondrial activity (indicated by yellowed mitochondria). Reduced G6PD activity leads to low NADPH levels, and GSH is depleted in favor of GSSG. Buildup of ROS leads to neuroinflammation, cell death, and kernicterus. BBB, blood-brain barrier.
The risk of kernicterus in G6PD deficient infants with TSB levels above 20 mg/dL (342 µmol/L appears to be comparable to that associated with Rh disease. • Thus, in the presence of G6PD deficiency, more aggressive treatment of these infants probably in indicated. Slusher TM, Vreman HJ, et al. Glucose-6-phosphate dehydrogenase deficiency and carboxy hemoglobin concentrations associated with bilirubin related morbidity and death in nigerian infants. J Pediatr 1995;126-102. Brows WR, Boon WH. Hyperbilirubinemia and kernicterus in glucose-6-phosphate dehydrogenase deficient infants in singapore. Pediatrics 1968;41:1055. Gibbs WN, Gray R, et al. G6PD deficiency and neonatal jaundice in jamaica. Br J Hematol 1979;43:263. Zahedpasha MD 2015
Comparison Between G6PD deficiencyin Icterin &Non-Icteric Neonates, Babol, Iran • 23.6%icteric AND 11.6%Nonicteric were G6PD deficient(p=0.0001) • 36%Icteric G6PD defAND18.2%Non-deficient icteric required Exchange Transfusion(p=0.0001). Zahed Pasha Y,J Medical Council I.R.Iran.2002 Zahed pasha J M,Councel Iran 2002
G6PD Enzyme Deficiency in Neonatal Pathologic Hyperbilirubinemia in Yazd. • 19 (18.1%) had G6PD deficiency, and consisted of 15 boys (29.4% of boys) and 4 girls (7.4% of girls). In 100% of cases, the jaundice began in the first week after birth. The average total serum bilirubin at hospitalization was 17.22 mg/dL. In 31.5% of the G6PD-defficient neonates, exchange transfusion became necessary, which is significantly more than the rate in • G6PD-sufficient(4.6%) neonates (P-value<0.05). PahlavanzadehM1, Hekmatimoghaddam S2, TeremahiArdestani M3, Ghafoorzadeh M4, Aminorraaya M5.Iran J Ped HematolOncol. 2013;3(2):69-72. Epub 2013 Apr 22.
Incidence, risk factors and causes of severe neonatal hyperbilirubinemia in the South of iran(Farsprovince). • More common causes of severe indirect hyperbilirubinemia were blood group incompatibility, G6PD deficiency, sepsis and unknown. Risk factors of severe hyperbilirubinemia were Male sex, previous siblings with severe hyperbilirubinemia, early discharge, NVD, Breast feeding and cultural background of mothers. NajibKS1, Saki F, Hemmati F, Inaloo S.Iran Red Crescent Med J.2013.
Zahedpasha Y, et al. Comparison of molecular mutations of G6PD deficiency gene between icteric and nonicteric neonates. Int J Mol Cell Med. 2013 Winter; 2(1): 14–20.
Zahedpasha Y, et al. Comparison of molecular mutations of G6PD deficiency gene between icteric and nonicteric neonates. Int J Mol Cell Med. 2013 Winter; 2(1): 14–20.
Comparison ofGilbert syndromein case and control grops of G6PD. d Neonates Zahedpasha Y, et al. Relation between Neonatal Icter and Gilbert Syndrome in Gloucose-6-Phosphate Dehydrogenase Deficient Subjects. J Clin Diagn Res. 2014 Mar; 8(3): 63–65
In many Asian, African, Mediterranean, and Middle Eastern countries, where G6PD deficiency is common, all newborns are screened for G6PD deficiency.
This screening has been associated with reduction of the instance of severe hyperbilirubinemia and kernicterus in several countries. In regions where G6PD deficiency is historically less common, an increase in global population movement has raised the question of whether G6PD-deficiency screening should be implemented everywhere. Watchko JF, Kaplan M, Stark AR, et al. Should we screen newborns for glucose-6phosphate dehydrogenase deficiency in the United States? J Perinatol 2013;33: 499–504.
HOW TO PREVENT KERNICTERUS? The incidence of kernicterus has risen in recent years because of a variety of factors: 1) infants are often discharged from the hospital within 24 to 48 hours of birth, despite the fact that TB levels often peak 4 to 5 days after birth; 2) the lack of proper monitoring at home allows the development of kernicterus 3)which might have otherwise been prevented if the infant were to remain at the hospital. Olusanya BO, Ogunlesi TA, Slusher TM. Why is kernicterus still a major cause of death and disability in low-income and middle-income countries? Arch Dis Child 2014;99:1117–21. Watchko JF. Identification of neonates at risk for hazardous hyperbilirubinemia: emerging clinical insights. PediatrClin North Am 2009;56:671–87. Ives K. Preventing kernicterus: a wake-up call. Arch Dis Child Fetal Neonatal Ed 2007;92:F330–1. Bhutani VK, Johnson L. Kernicterus in the 21st century: frequently asked questions. J Perinatol 2009;29:S2
Neonatal mortality in Europe and United States (EUROSTAT, CDC) Restoring Endogenous Glutathione: Activating Glucose-6-Phosphate Dehydrogenase Specifically, GSH depletion and increase in GSSG are hallmarks of bilirubin toxicity; a therapy directed at restoring the GSH/GSSG balance may be more helpful than general treatment with antioxidants. For example, pretreatment with N-acetylcysteine (NAC), a GSH precursor, reduced bilirubin toxicity in rat neuronal cells in culture. Vaz AR, Silva SL, Barateiro A, et al. Selective vulnerability of rat brain regions to unconjugated bilirubin. Mol Cell Neurosci 2011;48:82–93. Qaisiya M, Coda Zabetta CD, Bellarosa C, et al. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell Signal 2014;26:512–20.
Therefore, G6PD is the main driver of GSH regeneration. The loss of this major source of GSH in G6PD deficiency explains why higher rates and worse outcomes of kernicterus are observed in G6PDdeficient infants. Therefore, the authors propose a small-molecule activatoror chaperone of G6PD as a novel treatment for kernicterus. Discovery of molecular chaperones and activators is uncommon compared with the discovery of smallmolecule enzyme inhibitors, but a few small-molecule chaperones have been used successfully. Cunningham et al, Glucose-6-Phosphate Dehydrogenase Deficiency and the Need for a NovelTreatment to Prevent Kernicterus. Perinatology,2016
Glucose-6-Phosphate Dehydrogenase Deficiency and the Need for a Novel Treatment to Prevent Kernicterus
Mechanisms of pharmacologic chaperone or small-molecule activator. (A) A pharmacologic chaperone rescues misfolding. In the case of pharmacologic chaperones for acid b-glucosidase, the chaperone is competitively replaced by substrate. (B) A small molecule activator correcting a disordered region allosterically. (C) A small molecule activator binding in the active site, increasing productive interaction between substrate and catalytic residues.
In premature infant simple transfusions with G6PD-deficient red cells have been associated with hemolysis and severe hyperbilirubinemia requiringexchange transfusion . • Also massive intravascular hemolysis has occurred in an Indian neonate following an exchange transfusion with G6PD-deficient blood. //////////////////