Download

1 / 8
80 likes | 90 Views
This text discusses the characteristics and effects of different bonds in protein conformation, the folding and unfolding of helices, the role of disulfide bonds, ß-turns, motifs, and protein structure.

E N D
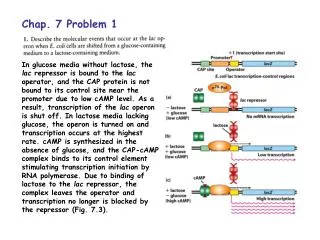
Chap. 4. Problem 1. Part (a). Double and triple bonds are shorter and stronger than single bonds. Because the length of a peptide bond more closely resembles that of a double bond, it suggests that a peptide bond has some characteristics of a double bond. Thus, a peptide bond is predicted to be stronger than a typical single bond. Part (b). The conclusion that a peptide bond resembles a double bond is further supported by the fact that the two -carbon atoms attached to the C-N peptide bond are always trans to one another. Rotation about this bond is apparently impossible. As shown in Fig. 4-2, peptide bonds have double bond character due to resonance of electrons among the atoms of the bond.
Chap. 4. Problem 4. The folding and unfolding of the Poly(Glu) and Poly(Lys) helices results from changes in the charge of the side-chains of these amino acids as a function of pH. As the pH of the solution containing Poly(Glu) is raised, the carboxyl group in the Glu side-chain dissociates and becomes negatively charged. This results in charge repulsion that destabilizes the helix. For Poly(Lys), elevation of pH causes the proton on the -amino group of the side-chain to dissociate. This neutralizes the charge on the R group and allows the peptide to fold into an helix. These transitions occur over a narrow range of pH due to the cooperative effects of R group ionization on structure along the length of the helix.
Chap. 4. Problem 5. Part (a). Disulfide bonds, being covalent bonds, have a strong effect on the stabilization of protein conformation. Disulfide bonds have marked effects on the mechanical strength (tortoise shell) and stiffness (wheat dough) of the proteins that contain them. Part (b). Heating of globular proteins leads to increased thermal motion of amino acid residues and disruption of many noncovalent interactions. However disulfide bonds are not broken by heating. The presence of these bonds prevents the polypeptide from becoming completely randomized and can facilitate the refolding of the protein when the temperature is reduced.
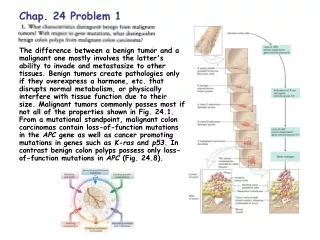
Chap. 4. Problem 7. Part (a). ß turns commonly (but not always) occur where Gly and Pro residues are found in the polypeptide chain. Thus, ß turns might occur at the residue 6-7 and 19-21 positions of the peptide shown. Note that Gly is found in ß turns because it is small and flexible. Pro is prevalent in ß turns because peptide bonds involving the imino nitrogen of Pro readily assume the cis configuration (Fig. 4-8). Part (b). An intrachain disulfide cross-linkage could occur between the Cys residues of the peptide located at positions 13 and 24. Part (c). On the basis of hydrophobicity (Table 3-1), Asp, Gln, and Lys might be expected to occur on the external surface of a globular protein. Ile and Ala would be more likely to occur in the nonpolar interior of a globular protein. Thr has intermediate polarity and might be present in either location.
Chap. 4. Problem 9. A motif (also called a fold) is a recognizable folding pattern involving two or more elements of secondary structure and the connections between them. A motif can be an elaborate structure involving multiple protein segments folded together, such as a large ß barrel. A domain is a part of a polypeptide chain that is independently stable or could undergo movements as a single entity with respect to the entire protein. Obviously, myoglobin is a complete three dimensional structure. In fact, it is all three of these terms. The folded structure (a.k.a. the globin fold) is a motif found in all globins. Myoglobin also consists of a single domain that can be referred to as the globin domain.
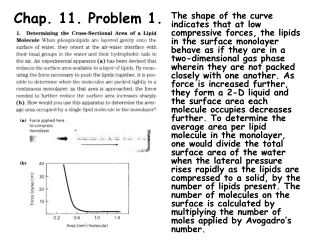
Chap. 4. Problem 10. The proteins shown in Panels (a) and (b) are dominated by ß and structure, respectively. Based on and angles, ß conformation occurs in the upper left quadrant of a Ramachandran plot, whereas -helical structure lies in the lower left quadrant (see Fig. 4-9). Thus it is most likely that the Ramachandran plot shown in Panel (c) belongs to the protein shown in Panel (a), and the plot in Panel (d) belongs to the protein shown in Panel (b).
Chap. 4. Problem 11. The sequence cleaved by this C. perfringens enzyme occurs in animal collagens (e.g., Gly-Pro-Y, see text p. 127). This suggests that the enzyme is a member of the collagenase family. This group of enzymes breaks down collagen and damages the connective tissue barriers of the skin and hide, etc. of the host animal allowing the bacterium to better infect the animal. Bacterial cells lack collagen, and thus the enzyme is not harmful to the bacterium itself.
Chap. 4. Problem 13. Based on visual inspection, Peptide b contains 3 Gly and 2 Pro residues, which are uncommon in helices. On the other hand, Peptide a contains 5 Ala residues which are frequently found in helices. These data suggest that Peptide a would be more likely to form an helix. This assumption is supported by calculating the values of ∆∆G˚ for folding of the residues of each peptide into an helix using the data in Table 4-2. For Peptide a, ∆∆G˚ for helix formation is 13 kJ/mol, whereas ∆∆G˚ is 41 kJ/mol for Peptide b. Because Peptide a has the lower value of ∆∆G˚, it is more likely to be folded into an helix. Note that ∆∆G˚ values are the differences in free energy change relative to alanine, that is required for an amino acid to take up the -helical conformation.