Download
1 / 11

E N D
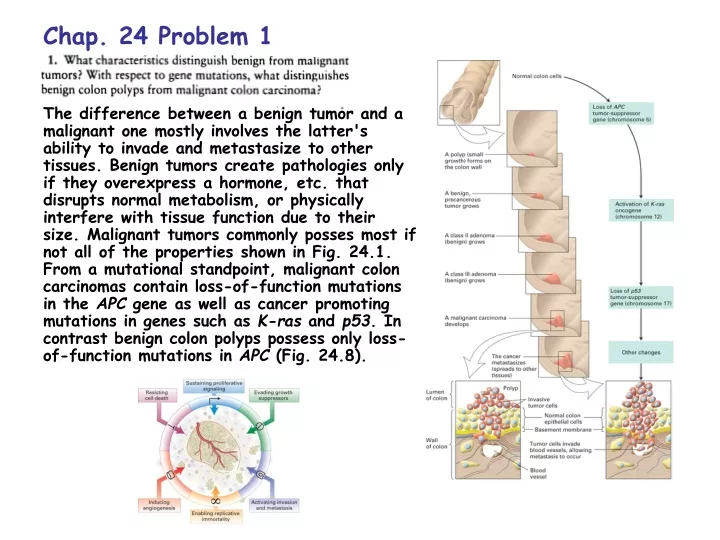
Chap. 24 Problem 1 The difference between a benign tumor and a malignant one mostly involves the latter's ability to invade and metastasize to other tissues. Benign tumors create pathologies only if they overexpress a hormone, etc. that disrupts normal metabolism, or physically interfere with tissue function due to their size. Malignant tumors commonly posses most if not all of the properties shown in Fig. 24.1. From a mutational standpoint, malignant colon carcinomas contain loss-of-function mutations in the APC gene as well as cancer promoting mutations in genes such as K-ras and p53. In contrast benign colon polyps possess only loss-of-function mutations in APC (Fig. 24.8).
Chap. 24 Problem 3 Malignant tumor cells secrete basic fibroblast growth factor (ßFGF), transforming growth factor (TGF, & vascular endothelial growth factor (VEGF) to recruit blood vessels for delivery of oxygen and nutrients to tumors. Tumors with their own vasculature can grow to a large size (Fig. 24.2a).
Chap. 24 Problem 6 The "multi-hit" model for cancer induction theorizes that metastatic tumor cells evolve from an original transformed cell via the accumulation of multiple mutations that increase its survivability and invasion potential. The multiple mutation theory is supported by the fact that the incidence of contracting most cancers increases steadily with age (Fig. 24.6). The multi-hit hypothesis also is supported by studies of the transformation of benign colon polyps into malignant colon carcinomas and by other research.
Chap. 24 Problem 7 Genes that control cell growth and proliferation are commonly mutated in cancers (Fig. 24.11). Gain-of-function mutations that increase the activity of growth-promoting signaling molecules (I), receptors (II), intracellular signal transduction pathways (III), or TFs (IV) are associated with cancers. These genes are referred to as proto-oncogenes. Commonly, only a single copy of the the gene needs to be altered. Loss-of-function mutations in tumor-suppressor genes such as cell cycle control proteins (V), DNA repair proteins (VI), or anti-proliferative factor receptors such as the TGFß receptor can cause cancer. Usually both copies of these genes need to be mutated. Lastly, gain-of-function mutations in anti-apoptotic genes and loss-of-function mutations in pro-apoptotic genes (VII) are associated with cancer. Based on the above considerations, ras, Bcl-2, MDM2, and jun are proto-oncogenes. p53 and p16 are tumor-suppressor genes.
Chap. 24 Problem 10 About 10% of human cancers have a hereditary basis. In most cases, the patient inherits one non-functional copy of a tumor-suppressor gene. Cancer is induced after the second functional copy of the gene is inactivated by mutation (loss of heterozygosity). Mutations in additional genes typically also are required. The induction of hereditary vs sporadic (spontaneous) retinoblastoma, which involves the RB gene, is compared in Fig. 24.13. The hereditary form of this cancer usually appears in childhood in both eyes. The sporadic form (which requires two somatic mutations) occurs later in life, and in only one eye. Hereditary retinoblastoma exhibits an autosomal dominant pattern of inheritance due to the fact that individuals with one mutant copy of RB have an increased probability of developing the disease.
Chap. 24 Problem 11 The concept of loss of heterozygosity (LOH) is explained in the preceding slide. In general, cells containing a predisposing loss-of-function mutation in one copy of a tumor suppressor gene are normal until a mutation inactivates the wild-type copy of the gene. Cancer cells commonly exhibit LOH in one or more tumor suppressor genes. As illustrated in Fig. 24.14a, non-disjunction (mis-segregation) events can result in LOH. Mutations that affect genes required for quality control at the spindle assembly checkpoint commonly are observed in cancers. This leads to nondisjunction events and LOH of tumor suppressor genes. Cancer cells commonly are aneuploid (contain aberrant numbers of chromosomes).
Chap. 24 Problem 12 After binding to hormones, growth factor RTKs such as the EGF receptor autophosphorylate themselves on cytosolic tyrosine residues. Phosphotyrosines then recruit proteins of signal transduction pathways to the receptor, activating signaling. Cytokine receptors are phosphorylated on cytoplasmic tyrosines by JAK kinases, leading to activation of signaling. (a) The viral protein gp55 binds to the erthropoietin receptor, activating JAK kinases in the absence of erythropoietin (Fig. 24.18). This leads to erythroleukemia. (b) In the Her2 receptor, the substitution of glutamine for valine in the transmembrane region of the receptor causes dimerization and activation of this growth factor receptor (Fig. 24.17). The resulting protein is known as the Neu oncoprotein, and is associated with some breast cancers.
Chap. 24 Problem 13 Ras signals via the MAP kinase pathway that is coupled to growth factor receptors. NF-1 (neurofibromatosis) is a GAP protein that catalyses GTP hydrolysis on Ras. Gain-of-function mutations in Ras increase signaling, whereas loss-of-function mutations in NF-1 activate signaling. Because only one copy of a gain-of-function mutation is needed to activate a process, mutations in Ras are more common than mutations in NF-1 in cancers. The first non-viral oncoprotein discovered was RasD. In RasD, amino acid substitutions at glycine-12 inhibit the GTPase activity of Ras keeping it locked in an active form. Constitutively activated RasD proteins occur in many bladder, colon, mammary, skin, and lung cancers, and in leukemias.
Chap. 24 Problem 14 The first oncoprotein discovered was the v-Src viral tyrosine kinase derived from c-Src (Fig. 24.19). c-Src is a member of a family of cytosolic tyrosine kinases implicated in cancers. In v-Src, the C-terminal 18 amino acids are deleted, including tyrosine-527. Phosphorylation on this tyrosine inactivates the wild-type c-Src protein. Because this regulatory site is missing from v-Src, the protein is constitutively active.
Chap. 24 Problem 15 Often growth-promoting TFs or signal transduction proteins are switched on by translocation of their genes to regions of the genome where they are highly expressed. In Burkitt's lymphoma, a translocation between the long arms of chromosomes 8 and 14 places the c-myc gene under the control of the enhancer for the antibody heavy chain gene (CH) (Fig. 24.22). This mutation is only associated with lymphomas because the antibody heavy chain gene is only expressed in B-lymphocytes. DNA containing a proto-oncogene can be amplified, leading to over-expression of the transforming gene product. The latter is illustrated in Fig. 24.12a for the N-myc oncogene. Staining shows that N-myc DNA in one copy of chromosome 4 is greatly amplified in human neuroblastoma cells. This type of mutation is not limited to lymphomas.
Chap. 24 Problem 16 TGFß is an anti-proliferative factor that signals via the Smad4 signal transduction pathway in cells such as pancreatic cells (Fig. 24.23). Loss-of-function mutations in Smad4 result in decreased expression of genes that limit cell proliferation. For example, the p15 gene is a tumor-suppressor gene that encodes an inhibitor of G1 cyclin-CDKs. p15 thus is important for slowing cell proliferation. The PAI-1 gene encodes an inhibitor of plasminogen activator. Plasmin degrades the extracellular matrix. The loss of the expression of these genes and other TGFß-controlled genes contributes to cell transformation.