Download

1 / 133
1.33k likes | 1.49k Views
Cystic Fibrosis - history. Dorothy Anderson 1938 - documented as clinical and pathological entity all dying at 1 week+: pneumonia, purulent bronchitis, bronchiectasis & abscess Blackfan -main problem is inspissiated secretions

E N D
Cystic Fibrosis - history • Dorothy Anderson 1938 - documented as clinical and pathological entity all dying at 1 week+: pneumonia, purulent bronchitis, bronchiectasis & abscess • Blackfan -main problem is inspissiated secretions • Fanconi - lack of pancreatic enzymes lead to malabsorption; association with bronchiectasis
CF - history • Faber 1943-45: the pathology of the pancreas is only one manifestation of the abnormality affecting all of the mucus glands • Harry Shwachman: diagnosis based on pancreatic enzyme analysis & clinical state • Anderson 1953 - abnormal sweat electrolytes
CF - history • Harvey White 1958 - Children’s Memorial Hospital Chicago • “Extensive antibiotic therapy and surgery will undoubtedly prolong life, so that we will see these children reaching their teens and young adult years with chronic pulmonary disease” • We have now moved beyond this with increasing numbers of patients in middle age
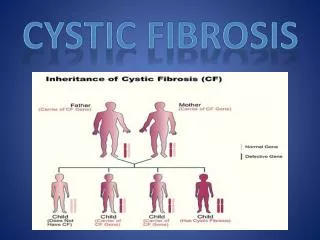
What is cystic fibrosis? • The most common genetic disease in caucasians • Gene frequency - carrier status 1 in 20-25 • Autosomal recessive inheritance • Live births in UK - approx 1 in 2000-2500 • A complex multisystem disease
How common ?Delta F508, G551D, G542X, 621+1(G>T) and 1898(G>A)
UK CF Survey • current population in UK 8000 to 9000 • About 250 children born per year with CF • About 150 patients with CF die each year • Incidence showing a fall from 1:2,500 (antenatal screening, cascade screening, increased ethnic mix) • Adult population > 50% total
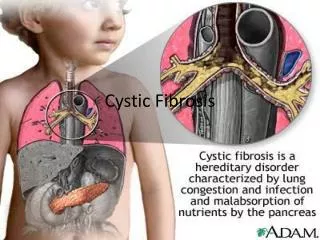
What is CF? • Affects respiration, digestion, hepatobiliary, reproductive, vascular, musculoskeletal, endocrine systems, and renal
CF • Multisystem disease but respiratory problems account for most of morbidity and mortality
What happens in CF? • The lungs are normal at birth • at post mortem - mucopurulent plugs, emphysema, collapse, pneumonia, haemorrhage • loss of cilia, bronchial epithelial metaplasia, massive neutrophil dominated inflammatory cell infiltrate
What is CF? • Over 90% pancreatic insufficient • sticky secretions to blocked ducts • secondary damage to secretory gland tissue • digestive juices - enzymes and bicarbonate • malabsorption, steatorrhoea, malnutrition, stunting, delayed puberty, fat sol vits
What is CF? • CF gene causes abnormal production of the CFTR protein • CFTR acts as a chloride channel in apical cell membrane and regulates sodium transport • CFTR found in lungs, salivary glands, sweat ducts, pancreas, liver, reproductive tract, kidney
CFTR • 1480 amino acids • Localizes to the apical cell membrane • Over 1500 mutations documented • ΔF508 is the most common mutation in Caucasian population • Five mutation classes
Normal I II III IV V Nosynthesis Block inprocessing Block inregulation Altered conductance Reduced synthesis Nonsense G542X MissenseG551D MissenseR117H MissenseA455E AA deletionDF508 Frameshift394delTT Alternativesplicing3849+10kbC→T Splice junction1717-1G→A Molecular Consequences of CFTR Mutations CFTR Vankeerberghen A, et al. J Cyst Fibros. 2002. http://www.genet.sickkids.on.ca/cftr/
Pathophysiology • CFTR expressed on the luminal surface of epithelial cells • Impairment of function • Defective chloride transport • Increased sodium transport • Increased fluid reabsorption • Overall increased viscosity of epithelial surface secretions
glycosylation Golgi apparatus mRNA Endoplasmic reticulum The airway cell- normal epithelial cell - Cl- wild type CFTR Cl- Cl- Cl- Cl- ATP Chromosome 7 800 different mutations
Impairment of CFTR function causes reduced fluid production. Enhanced sodium absorption through epithelial Na+ channels (ENaC) and basolateral Na/K ATPase pumps results in increased fluid absorption leading to dryer airways and impaired ciliary clearance
Rare presenting features • Nasal polyps/sinus disease - 5% • rectal prolapse - 2% • oedema - 2% • other - male infertility, pancreatitis
At time of diagnosis • CF is a serious disorder despite major advances in Rx and understanding • Health now and longterm depend on effective Rx • Outlook improves year by year • Complex illness needing specialist care • Sweat test sibs, “cascade screen”
CF - survival • 1938 - 70% died in the 1st year • 2000 - 50% survive to over 30 years • Median age death 1987 to 1999, 19 to 23 yrs • Median predicted survival 1999 = 29 yrs [25.5 in 1985] • Adult [>17 yrs] USA - 1989 & 1999 31% & 38% of all patients • 2007 average survival is 39 years
MEDIAN SURVIVAL AGE FOR PATIENTS WITH CF AT VARIOUS TIMES SINCE FIRST DESCRIPTION OF CF
Increasing survival • >50% patients now adults (UK data) • Median survival 38.6 years (USA data)
100 90 80 Percent predicted FEV1 70 60 1990 2001 50 40 6 8 10 12 14 16 18 24 26 28 30 20 22 Age (Years) Median Percent Predicted FEV1 vs Age: 1990 and 2001 Cystic Fibrosis Foundation. Patient Registry Annual Data Report. 2002.
Pseudomonas aeruginosa • Wilmott 1985 • Chronic infection in 1st 5 years - 20% survival to 16 years • No infection - 95% survival to 16 years
Survival with CF • Many reasons for improved prognosis BUT • survival better without chronic chest infection • USA data 1990s: average survival with chronic PA infection = 28 years BUT without chronic PA infection = 39 years
Pseudomonas aeruginosa infection - Kerem 1990 • Life after pseudomonas in 895 patients attending Toronto clinic - At 7 years of age the mean percentage predicted FEV1 was 10% lower in children already infected compared with those who were not - p<0.01 • Throughout clinic population FEV1 in those with PA was about 10% less of predicted than those without PA
Chronic Pa Endobronchitis Is Associated with Poor Survival in CF 1.0 Pa-negative(n = 12) 0.9 Nonmucoid Pa (n = 19) 0.8 Mucoid Pa (n = 50) 0.7 Fraction surviving 0.6 0.5 0.4 0.0 20 50 60 70 80 90 100 0 40 10 30 Time (months) Henry RL, et al. Pediatr Pulmonol. 1992.
Symptoms of infection • Cough one of earliest and most important • lethargy • weight loss • fever • more sputum, white to yellow or green • dyspnoea
Age-specific prevalence of infections in CF patients 2003 Cystic Fibrosis Patient Registry 2003 – Annual Data Report. October 2004.
Disease begins in the small airways Bronchioles fill with chronic mucopurulent material Childhood and adolescence: progressive loss of small airway function Pathology of CF Lung Disease At birth: lungs are normal Tomashefski JF, et al. The pathology of cystic fibrosis. In: Cystic Fibrosis. 1993.
Structural Changes Occur Even in Mild CF Lung Disease • 60 children with CF (6-12 years old) • Mean FEV1 = 102% predicted • 11/37 patients with normal lung function (all PFT values >85% predicted) had HRCT evidence of bronchiectasis • FEV1: 91% of predicted • FVC: 95% of predicted • FEF25-75: 89% of predicted Brody AS, et al. J Pediatr. 2004.
CT Scans of Normal and CF Lungs Normal CF Tiddens HA. Pediatr Pulmonol. 2002. http://www.rrcc-online.com/paprogram/HISTHTML/RADNORM/CHCT02.HTM.
The Vicious Cycle of CF Lung Disease • Ibuprofen • Corticosteroids Inflammation Obstruction • Airway clearance • Dornase alfa • Hypertonic saline • Bronchodilators Infection • Tobramycin • Azithromycin Chmiel JF, et al. Clin Rev Allergy Immunol. 2002.
Treatment of the chest in children with CF - 1st principle • PREVENTION • PREVENTION • PREVENTION
Recovered from toys, baths, soaps, nebulizers, sink drains, and hands of patients and staff Nonmucoid strains can survive for 24 hours, mucoid strains for ≥48 hours On dry surfaces, pathogens suspended in sputum can live for up to 8 days Pseudomonas aeruginosa: Sources in the Healthcare Environment Saiman L. et al. Clin Microbiol Rev. 2004.
P aeruginosa: Patient-to-Patient Transmission • Shared strains found in Germany, United Kingdom, Australia, Denmark, and United States • Linked to antibiotic resistance • May involve transmission from Pa-positive to Pa-negative patients • Documented between siblings with CF • Linked to summer camps • Possibly via contaminated environment Saiman L. et al. Clin Microbiol Rev. 2004.
Modes of Transmission • Direct: body-to-body surface transfer • Kissing • Touching • Patient care • Shaking hands • Indirect: contact with a contaminated object • Toys • Shared toothbrushes • Eating utensils • Gloves • Respiratory equipment Saiman L. Cystic Fibrosis Foundation [webcast]. 2004.