Download
1 / 1
30 likes | 139 Views
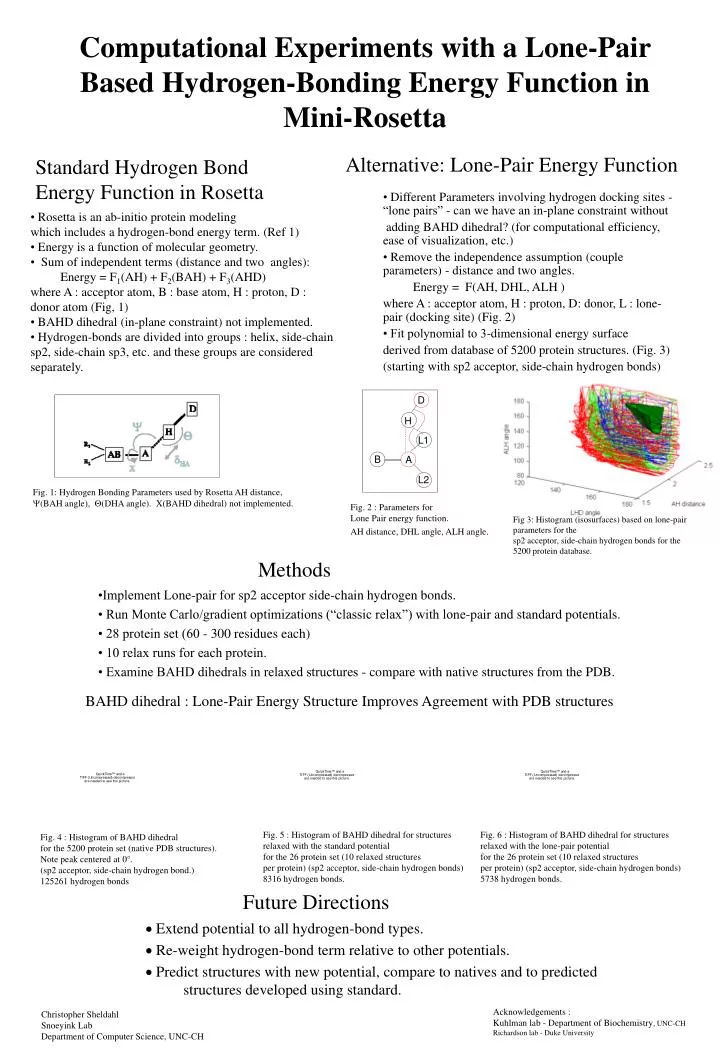
D. H. B. A. L2. L1. Computational Experiments with a Lone-Pair Based Hydrogen-Bonding Energy Function in Mini-Rosetta. Alternative: Lone-Pair Energy Function. Standard Hydrogen Bond Energy Function in Rosetta.

E N D
D H B A L2 L1 Computational Experiments with a Lone-Pair Based Hydrogen-Bonding Energy Function in Mini-Rosetta Alternative: Lone-Pair Energy Function Standard Hydrogen Bond Energy Function in Rosetta • Different Parameters involving hydrogen docking sites - “lone pairs” - can we have an in-plane constraint without • adding BAHD dihedral? (for computational efficiency, ease of visualization, etc.) • Remove the independence assumption (couple parameters) - distance and two angles. • Energy = F(AH, DHL, ALH ) • where A : acceptor atom, H : proton, D: donor, L : lone-pair (docking site) (Fig. 2) • • Fit polynomial to 3-dimensional energy surface • derived from database of 5200 protein structures. (Fig. 3) (starting with sp2 acceptor, side-chain hydrogen bonds) • Rosetta is an ab-initio protein modeling program • which includes a hydrogen-bond energy term. (Ref 1) • Energy is a function of molecular geometry. • Sum of independent terms (distance and two angles): Energy = F1(AH) + F2(BAH) + F3(AHD) where A : acceptor atom, B : base atom, H : proton, D : donor atom (Fig, 1) • BAHD dihedral (in-plane constraint) not implemented. • Hydrogen-bonds are divided into groups : helix, side-chain sp2, side-chain sp3, etc. and these groups are considered separately. Fig. 1: Hydrogen Bonding Parameters used by Rosetta AH distance, (BAH angle), (DHA angle). X(BAHD dihedral) not implemented. Fig. 2 : Parameters for Lone Pair energy function. AH distance, DHL angle, ALH angle. Fig 3: Histogram (isosurfaces) based on lone-pair parameters for the sp2 acceptor, side-chain hydrogen bonds for the 5200 protein database. Methods •Implement Lone-pair for sp2 acceptor side-chain hydrogen bonds. • Run Monte Carlo/gradient optimizations (“classic relax”) with lone-pair and standard potentials. • 28 protein set (60 - 300 residues each) • 10 relax runs for each protein. • Examine BAHD dihedrals in relaxed structures - compare with native structures from the PDB. BAHD dihedral : Lone-Pair Energy Structure Improves Agreement with PDB structures Fig. 5 : Histogram of BAHD dihedral for structures relaxed with the standard potential for the 26 protein set (10 relaxed structures per protein) (sp2 acceptor, side-chain hydrogen bonds) 8316 hydrogen bonds. Fig. 6 : Histogram of BAHD dihedral for structures relaxed with the lone-pair potential for the 26 protein set (10 relaxed structures per protein) (sp2 acceptor, side-chain hydrogen bonds) 5738 hydrogen bonds. Fig. 4 : Histogram of BAHD dihedral for the 5200 protein set (native PDB structures). Note peak centered at 0°. (sp2 acceptor, side-chain hydrogen bond.) 125261 hydrogen bonds Figure 2: Gary Bishop giving a head-mounted display demo to UNC System President Molly Broad. (Arial 24 pt italic) Future Directions Extend potential to all hydrogen-bond types. Re-weight hydrogen-bond term relative to other potentials. Predict structures with new potential, compare to natives and to predicted structures developed using standard. Acknowledgements : Kuhlman lab - Department of Biochemistry, UNC-CH Richardson lab - Duke University HERE Christopher Sheldahl Snoeyink Lab Department of Computer Science, UNC-CH YOUR NAME HERE (Arial 28 pt italic) YOUR PROJECT URL HERE