Download

1 / 27
280 likes | 749 Views
Sickle Cell, Macrocytic & Microcytic Anemia. Primary care II Melisa Dervisevic Ronald Otwori. Definition.

E N D
Sickle Cell, Macrocytic & Microcytic Anemia Primary care II Melisa Dervisevic Ronald Otwori
Definition • Sickle Cell Anemia: disease of the erythrocytes caused by an autosomal recessive single gene defect in the B-globin chain of adult hemoglobin (HbA) that produces a mutant form of hemoglobin known as sickle hemoglobin (HbS). In can occur either from homozygosity for the sickle mutation in the beta globin chain of hemoglobin (HbSS) or from compound heterozygosity of a sickle beta globin mutation with another beta globin mutation (e.g., sickle-beta thalassemia) • Macrocytic anemia: mean corpuscular volume (MCV) greater than 100fL. Red blood cells are larger than normal • Microcytic anemia: MCV less than 80fL. Red blood cells are smaller than normal (First consult,2013 & Schrier, 2013)
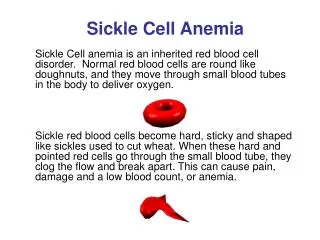
Pathophysiology • Sickle Cell Anemia: HbS results from the substitution of a valine for glutamic acid as the sixth amino acid of the beta globin chain, which produces a hemoglobin tetramer (alpha2/beta S2) that is poorly soluble when deoxygenated. The polymerization of deoxy HbS is important to vaso-occlusive phenomena. The polymer assumes the form of an elongated rope-like fiber, aligning itself with other fibers, resulting in distortion into the classic crescent or sickle shape and a marked decrease in red cell deformability. • Macrocytic anemia: Can be classified as megaloblastic and nonmegaloblastic. Megaloblastic processes are characterized on the peripheral smear by macro-ovalocytes and hypersegmented neutrophils, absent in nonmegaloblastic. Nonmegaloblastic processes have round macrocytes or macroreticulocytes. • Microcytic anemia: depletion of iron stores. (Kaferle & Strzoda, 2009, Vichinsky, 2013)
Etiology • Sickle cell anemia • Inherited condition • If both mother and father have sickle cell trait and each carry one copy of the mutated hemoglobin S gene, their offspring have ¼ chance of getting homozygous sickle cell anemia • If one parent is homozygous (HbSS) and the other parent is heterozygous (HbS), the risk of affected offspring is 50% • If both parents are HbSS, all offspring will be affected • Homozygous for the HbS gene will develop sickle cell anemia (spend their live with sickle cell crisis and will die) • Heterozygous for HbS are carriers of the sickle cell trait and will not have painful sickle cell crisis (First consult, 2014)
Etiology Macrocytic anemia Microcytic anemia • Alcohol • Vitamin B12 deficiency • Folate deficiency • Medications • Bone marrow dysplasias • Reticulocytosis • Iron-Deficiency • Blood loss • Anemia of chronic disease • Lead poisoning • Thalassemia • Sideroblastosis (Kaferle & Strzoda, 2009, Dunphyet al, 2011)
Incidence Sickle cell anemia Macrocytic/Microcytic • 4,000-5,000 pregnancies are at risk for sickle cell disease each year in U.S • 6-9 million infants are born each year with sickle cell disease in Africa • Sickle cell anemia accounts for 60-70% of sickle cell disease in the U.S. • Sickle cell disease occurs in 1 in 600 African-American infants • 8-10% of African-American neonates in the U.S. are carriers of the sickle cell trait • Highest prevalence among people of African, African-American, Mediterranean, Middle Eastern, East Indian, Caribbean, and Central or South American • 3% of the general population have macrocytosis • 40% of macrocytic anemia is found amongst the cases of severe anemia in pregnancy • Prevalence is greatest among people of northern Europeans and Caucasian (Macro) • Macro: Incidence increases past age 60 • Micro: Incidence highest among women with childbearing age. Up to a third of pregnant women develop anemia in the 3rd trimester • Micro: In the U.S. 20% of adult women are affected and 3% of adult men • Premenopausal women and children are at greatest risk for iron deficiency anemia (First consult, 2014, Kaferle & Strzoda, 2009, Domino,2010)
Screening/Risk Factors • Sickle cell anemia: • Newborn screening- two separate blood tests. First blood sample is obtained by heel stick or cord blood to confirm that the child has the abnormal hemoglobin. Second test is the Confirmatory test performed by using hemoglobin electrophoresis and/or deoxyribonucleic acid (DNA) to confirm the type of sickle cell disease (blood smear) • Complete blood count and reticulocyte count • Macrocytic/Microcytic anemia: • Routine complete blood count • History and physical (Vichinsky & Mahoney, 2013, Dunphy et al, 2011)
Clinical Findings • Sickle cell anemia: • Infants- usually asymptomatic until 4 months of age, when hemolytic anemia becomes obvious. Moderate anemia by 6-12 months of age or with more serious crisis and pain. Painful swelling of the hands and feet, known as hand-foot syndrome or dactylitis, is often the first presentation • Clinical presentations vary from patient to patient due either to the acute-occlusive consequences of sickle cells or chronic hemolysis and resultant anemia. Some individuals may remain asymptomatic into late childhood. • Acute s/s: acute chest pain and fever, persistent pain (usually in the skeleton, chest, and/or abdomen), malaise, cough, diarrhea and or vomiting, dactylitis, abdominal discomfort, stroke ( affects 10% of children), sudden neurologic deficits ( difficulty with language, writing, and/or reading, seizures, sensory deficits), pallor, temp >101.3F, tachycardia, dyspnea, limited ROM, splenomegaly, and possible shock • Chronic s/s: excessive fatigue, pain ( usually back and hips), breathlessness on exertion, decreased exercise tolerance, growth and development delay, jaundice, visual disturbances, hepatomegaly/splenomegaly, pallor, cardiomegaly with grade II-III systolic ejection murmur, bone deformities, leg ulcers. (First consult, 2014)
Clinical Findings Macrocytic Microcytic • Mild anemia may be asymptomatic but might have s/s if there are underlying disease • Shortness of breath on exertion • Fatigue • Palpitations • Exacerbation of angina • Pallor • Bounding pulse • Weakness and fatigue • Decrease in appetite • Changes in texture of nails, they become fragile and break easily • Severe headaches and difficulty concentrating • Tachycardia • Shortness of breath • Dizziness • Chest pain (Draper, 2011, medicalhealthtests, 2012)
Differential Diagnosis Sickle cell anemia Macro/Micro anemia • Anemia resulting from another cause- sickle thalassemia or hemoglobin C disorders • Gout • Septic arthritis • Connective tissue disease • Avascular necrosis • Acute abdominal disorders ( RUQ pain ? Acute cholelithiasis, hepatic crisis, pancreatitis) • Osteomyelitis • Congenital Syphilis • Megaloblastic: atrophic gastritis, enteral malabsorption, HIV treatments, primary bone marrow disorders, inherited disorders • Nonmegaloblastic: alcohol abuse, medication side effects, myelodysplasia, hypothyroidism, liver disease, hemolysis, hemorrhage, COPD • Micro: Iron-deficiency anemia, anemia of chronic disease, thalassemia, and sideroblastic anemia (Firstconsult, 2014, Kaferlee & Strzoda, 2009)
Social/Environmental Considerations • Sickle cell anemia- to prevent or reduce long term complications the individual should: have regular physical exams every 3-6months, regular eye exams, have adequate rest, warmth and increased fluid intake, avoid crowds (increase chance of infection), avoid excess demands on the body that would increase oxygen needs (physical overexertion or stress), and do not smoke/avoid secondhand smoke • Macrocytic/Microcytic: Include protein and iron-containing foods (meat, beans, and leafy green vegetables) in the diet, increase fluid intake, limit tea, coffee, and caffeinated beverages, take iron with orange juice or ascorbic acid to increase absorption (Domino, 2010)
Laboratory Tests/Diagnostic Sickle cell anemia Macro/Micro anemia • CBC and reticulocyte count: used to evaluate the number and quality of erythrocytes, hemoglobin content and leukocyte count • Peripheral blood smear: erythrocyte morphology • Hemoglobin solubility: screen for HbS • Hemoglobin studies: differentiate between heterozygosity and homozygosity • DNA analysis: confirm the presence of sickle cell disease or sickle cell trait • Iron studies: help determine whether iron deficiency is the cause • Macro: CBC, peripheral smear, reticulocyte count, vitamin B12 serum level. Also consider CMP, TSH, methymalonic acid and homocysteine levels • Micro: Hemoglobin and hematocrit level, MCV level, ferritin, transferritin saturation, total iron-binding capacity, CBC, peripheral smear, reticulocyte count (Firstconsult,2014, Kaferle & Strzoda, 2009)
SCD Management • Pharmacologic • Prevention of infection: Prophylactic PCN should be given between 2 months-5years to prevent serious infection such as pneumonia • Pain management: Acetaminophen (with or without codeine) or NSAIDs are used for mild pain. Stronger opioids (hydrocodone) used for moderate pain. If the pain is severe, parenteral opioid may be required (morphine is preferred) • Blood transfusion: acute symptomatic exacerbations of anemia (SOB or chest pain) • Hydroxyuea: increases the production of HbF, HbS cannot polymerize and decreases the sickling of the erythrocytes • Erythropoietin: patients who develop end-stage renal disease • Fluid replacement: mild dehydration. Fluid and electrolyte balance is often disrupted during pain crisis and infections • Non-Pharmacologic • Transcutaneous electrical nerve stimulation (TENS) • Warm compresses • Physical therapy • Hypnotherapy • Relaxation therapy • Avoid being chilled (increases pain) • Increase fluids by mouth • Avoid constipation • Nutritional supplements- protein and iron (Firstconsult, 2014)
Macro/Micro Management • Correct the deficiency that has caused the macrocytosis or microcytosis, with or without anemia. (e.g., vitamin B12/folate replacement if macro or ferrous sulfate 325mg oral TID if micro) • Treat the underlying condition that led to the deficiency (e.g., if hypothyroidism, treat that) • Liquid iron are useful in children (3mg/kg/d) • Foods and beverages containing ascorbic acid (vitamin C) should be encouraged to increase iron absorption • Parenteral iron therapy should be initiated if Hgb <6g/dl, malabsorption, if higher oral doses and use of vitamin C fails (Domino, 2010)
SCD Complications • Acute painful episodes: previously called sickle cell crisis. Most common type of vasoocclusive event. • Neurological: The Cooperative study of SCD found that 24% of individuals with sickle cell anemia experienced a stroke by 45. In childhood, 25% have silent ischemic lesions that may impair neurocognitive function. Increased risk for intracranial hemorrhage. 2-3 times more likely to have epilepsy. • Psychosocial issues: low self-esteem, social isolation, poor family relationships, withdrawal, anxiety, depression, and inappropriate pain coping strategies. • Reduced or absent splenic function: due to splenic infarction secondary to sickling of red cells within the spleen • Infection: caused by inability of spleen to filter microorganisms. Bacteremia caused by S.pneumonia or H. influenza type B is common. Meningitis primarily in infants and young children. Bacterial pneumonia caused by mycoplasma, chlamydia, and legionella. Osteomyelitis caused from infection of infarcted bone • Bone: bone infarction and necrosis due to accelerated hematopoiesis • Cardiac: increased cardiac output and acute MI. • Dermatology: vasoocclusion in the skin causes leg ulcers • Hepatobiliary: hepatic dysfunction caused by acute hepatic ischemia, benign cholestasis, transfusion iron overload, cholelithiasis, drug toxicity • Pulmonary: pulmonary arterial circulation has low oxygen tension and low pressure. O2 sat is below normal. Acute chest syndrome, include pneumonia, infarction due to in situ thrombosis, and embolic phenomena due to fat embolism and bone marrow infarction. • Renal: 18% have renal failure. • Retinopathy: retinal artery occlusion, retinal detachment and hemorrhage can occur • Urology: prolonged priapism (erection lasting longer than four hours). 80% of males will experience before age 18 • Anemia: acute severe anemia, aplastic crisis, hyperhemolytic crisis • (Rogers, 2013)
Macro/Micro Complications • Neglecting to identify hidden bleeding points, especially a bleeding malignancy • Maternal iron deficiency negatively affects mother-child interactions. Iron supplementation protects against these negative effects • Hemolysis (rapid fall in hemoglobin concentration, reticulocytosis, and/or abnormally shaped RBC in the absence of blood loss • Intravascular hemolysis • Abnormal cells in the circulation (such as blast forms). Worry about malignancy (Dunphy et al, 2011)
Follow-up Sickle cell anemia Macro/Micro anemia • Follow-up should be every 3-6 months if they have not been in crisis • Frequency and duration of crisis may increased the frequency of follow up visits. • Laboratory evaluations should occur periodically (CBC, fasting blood sugar, electrolytes, renal and liver function studies, and urinalysis) • Annual or bi-annual 12-lead ECG • Every patient should be evaluated by hematologist twice per year • Retinal examinations should be performed twice a year • Screening for pulmonary HTN • Yearly screening for proteinuria • Macro: Follow up with vitamin B12 deficiency depends on the severity. CBC and vitamin B12 levels should be done monthly after starting oral therapy. If parenteral therapy is required, then follow up should be every 2 weeks. LFT’s should be monitored. • Micro: Iron-deficiency anemia that is mild should be followed every 4-6 months. Anemia of chronic disease should be followed monthly for the first 6 months if getting erythropoietin-alpha injections. (Domino, 2010, Firsconsult, 2014)
Counseling/Education • Sickle cell anemia • Parents should be given information about sickle cell disease as early as possible • Importance of regularly scheduled health maintenance visits: PCN prophylaxis and immunizations, including pneumococcal vaccination • Information should be given regarding home management of pain, importance of adequate hydration, and avoidance of heat/cold • Parents should be taught how to recognize s/s that require medical attention such as enlarged spleen, fever, pallor of skin, lips, nailbeds, any respiratory sxs, pain, or inability to move extremities • Information should be given on complications • Adolescents should get information about contraception, carrier testing of partners, genetic counseling, and prenatal diagnosis • Macro: Patients need to be taught how to include folic acid and vitamin B12 in their diet. They should increase dark-green vegetables in one or two meals daily. They should be educated on basic s/s of macrocytic anemia so they can self-monitor • Micro: Education should be focused on self-care and primary care management of the underlying cause. Iron-deficiency anemia- patients should take ferrous sulfate on an empty stomach, eat foods that are rich in iron, vitamin C, and B-complex vitamins, remain active, rest when needed, and self-monitor for any fatigue, SOB, pale-colored stools, or tachycardia. (Firstconsult, 2014, Domino, 2010)
Consultation/Referral • Sickle cell anemia: Anesthesiology (perioperative care and pain management), Cardiology (pulmonary HTN), Endocrine (poor linear growth and delayed puberty), Nephrology (HTN, proteinuria, or renal insufficiency, Neurology (stroke, silent infarct, or headache), Orthopedics (avascular necrosis), Pulmonary (asthma or pulmonary HTN), Surgery (cholelithiasis or splenectomy), Urology (prolonged priapism), Psychologist (individual counseling) • Macro: Refer to hematologist if there are neurological symptoms, if patient is pregnant, or the suspected cause is hematological malignancy. Refer to gastroenterologist if there is malabsorption of vitamin B12 or folate is suspected, if the person has pernicious anemia or GI sx, if there is suspicion of gastric cancer, or the patient is folate deficient. • Micro: Refer to Gastroenterology if GI malignancy is suspected or other type of occult blood loss. Upper and lower endoscopy might need to be done. Hematologist referral if the cause of anemia is thalassemia or sideroblastosis (Dunphy et al, 2011, Rogers, 2013)
Questions • 1. If both the mother and father carry a sickle cell trait and each carry a copy of the hemoglobin S gene, what is the chance that the child will have homozygous sickle cell anemia? • 50% • 25% • 75% • 0% • 2. Sickle cell anemia has the highest prevalence among what people? • African • Middle Eastern • Chinese • Caribbean • 3. The first clinical presentation of an infant with sickle cell anemia is? • Respiratory distress • Diarrhea • Feeding difficulty • Swelling of hands and feet (dactyltis)
4. When an individual is homozygous for the hemoglobin S gene in sickle cell anemia, they will? • Not experience sickle cell crisis • Not carry or have the disease • Spend their whole life in sickle cell crisis • Not have any symptoms • 5. Non-pharmacologic management of sickle cell disease include all of the following except? • Physical therapy • Warm compresses • Iron supplements • Relaxation therapy • 6. The major cause of macrocytic anemia is? • Iron deficiency • Vitamin B12 and folate deficiency • Alcohol • Medication
7. The number one cause of microcytic anemia is? • Iron deficiency anemia • Vitamin B12 deficiency • Blood loss • Anemia of chronic disease • 8. Dietary education for a patient that has microcytic anemia should include what? • Increase the amount of fiber • Eat foods rich in iron and vitamin C • Increase the amount of fruit intake • Avoid spicy and fried food
9. The two main referrals that should be made in micro/macro anemia are? • Pulmonology and cardiology • Gynecology and Gastroenterology • Hematology and Cardiology • Gastroenterology and Hematology • 10. In order to prevent infection with sickle cell disease, what management is done? • Penicillin is given prophylactically from 2 months-5 years • Levaquin is given 2 years after diagnosis • Good handwashing • Doxycycline is given when diagnosed for a year
References • Domino, F.J. (2010) The 5-minute clinical consult. United States: Lippincott Williams & Wilkins • Draper, R. (2011) Macrocytosis and macrocytic anemia. Retrieved from http://www.patient.co.uk/doctor/macrocytosis-and-macrocytic-anemia • Dunphy, L.M., Winland-Brown, J.E., Porter, B.O. & Thomas, D.J. (2011) Primary care the art and science of advanced practice nursing. Philadelphia, PA: F.A. Davis Company • First consult (2013) Sickle cell disease. Retrieved from http://www.mdconsult.com/das/pdxmd/body • Kaferle, J. & Strzoda, C.E. (2009) Evaluation of macrocytosis. American Family Physician, 79(3), 203-208. • Medical health tests (2012) Microcytic anemia: causes, symptoms, and treatment. Retrieved from http://www.medicalhealthtests.com/diseases-and-tests/anemia/microcytic-anemia.html • Rogers, Z.R. (2013) Routine comprehensive care for children with sickle cell disease. Retrieved from http://www.uptodate.com/contents/routine-comprehensive-care-for-children-with-sickle-cell
Schrier, S.L. (2013) Approach to the adult patient with anemia. Retrieved from http://www.uptodate.com/contents/approach-to-the-adult-patient-with-anemia • Vichinsky, E.P. (2013) Overview of the clinical manifestations of sickle cell disease. Retrieved from http://www.uptodate.com/contents/overview-of-the-clinical-manifestations-of-sickle-cell-disease • Vichinsky, E.P. & Mahoney, D.H. (2013) Diagnosis of sickle cell disorders. Retrieved from http://www.uptodate.com/contents/diagnosis-of-sickle-cell-disorders