Download

1 / 57
620 likes | 1.13k Views
Sickle Cell Anemia. Emergency management of acute neurologic events in children Dr. Mary-Lou O’Byrne September 25, 2003. OUTLINE. Case presentation Sickle Cell Anemia fundamentals Genetics Pathogenesis “Crises” Sickle Cell Disease Care Consortium guidelines Therapies Prevention.

E N D
Sickle Cell Anemia Emergency management of acute neurologic events in children Dr. Mary-Lou O’Byrne September 25, 2003
OUTLINE • Case presentation • Sickle Cell Anemia fundamentals • Genetics • Pathogenesis • “Crises” • Sickle Cell Disease Care Consortium guidelines • Therapies • Prevention
CASE (C.F.) • 6 ½ year old black Canadian with known Sickle Cell Disease • Well for 3 years • Prior hx of splenic sequestration events • Admitted to PLC x 3 in first 3 years of life • Meds: Pen V and Folate • Immunizations: UTD
Presentation (C.F.) • Well at bedtime • @ 23:00 woke crying with bilaterally painful legs • Mother is rubbing her legs when pt stops crying, stiffens, and has a large emesis • Generalized tonic seizure x 5 minutes • EMS called
On arrival in E.D. • Eyes closed but will open eyes to voice commands • c/o headache and abd pain • Oriented to place and self • PERL, marked nystagmus, diplopia on lateral gazes w/ R>L lat rectus palsy • Ataxia, unable to sit • Vomiting with any mvmt • Marked dysmetria • DTR symmetric and brisk, Plantars downgoing
Exam cont’d • Chest: clear, sats 85% in R/A, RR: 20 • CVS: HR 60-80, murmur noted • ABD: liver 4 cm down, Spleen not palp • MSK: no tenderness, swelling or erythema
Investigations • CXR: mild cardiomegaly • Hgb 73 • WBC 16.0 => 9 to 12 in hospital • Plt 529 • CBG: pH 7.41 pCO2 38 pO2 191 • Lytes N INR 1.2 PTT 23 • Type and screen done • CT head done immediately and “normal”
Treatment in E.D. • IV normal saline at maintenance • O2 • ICU consulted • Admitted onto clusters w/in 2 hours of arrival
Course in Hosp • Resolution of most of her neurologic findings within a few hours • I phoned neurology after my shift and they had not been consulted yet • Hematology consulted later in day • ECHO and ECG confirmed mild rt and left ventricular enlargement and norm ventricular func • MRI/MRA done and abnormal
Course in Hospital • Exchange transfusion in ICU on fourth day • Sickle cells 84% to 34% • Residual left cerebellar signs on discharge • Follow up w/ brain injury team, hematology for ‘possible’ chronic transfusion Rx
Sickle Cell Anemia • Hereditary hemoglobinopathy • 8% of black Americans heterozygous for HbS • 1910 Herrick first described ‘sickled’ cells • Heterozygote (40% HbS) protects against falciparum malaria • African descent > Hispanic, Arabians, Indians and whites • 1973 life expectancy = 14.3 years • 1994 life expectancy = 42 years men / 48 years women • Most American states screen all newborns • (1:625 Afro-American births)
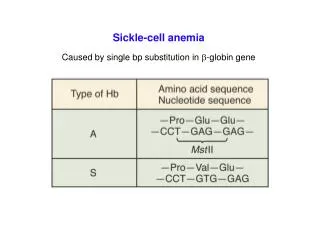
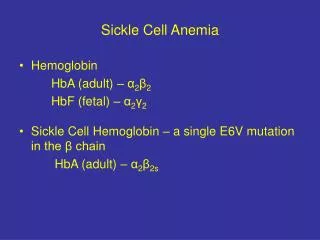
Sickle Cell Anemia • Structurally abnormal hemoglobin • Normally tetramer of 4 globin chains: • 96%HbA (alpha2 beta2) • 3% HbA2 (alpha2 delta2) • 1% HbF (alpha2 gamma2) • SCD: HbS is abnormal Beta chain • Valine replaces glutamine on beta-globin • Charge at site altered, allows polymerization of Hb • Copolymerization w/ HbS>C>D etc
Pathogenesis • On deoxygenation HbS aggregates and polymerizes (like viscous gel) • Sickling is initially reversible w/ O2 • HbS returns to depolymerized state • Recurrent sickling => memb damage and irreversible sickling • RBCs lose K+ & H2 O and gain Ca++ • Dehydrated cells
Pathogenesis - 2 • HbF (fetal = alpha2 gamma2) inhibits polymerization of HbS • Newborns protected until 5-6 months • Coexistent alpha-thalassemia is protective b/c it reduces the conc of Hb in a RBC • Acidosis reduces O2 affinity of Hb and deoxygenates HbS • HbS causes: 1)chronic hemolytic anemia 2)occlusion of small blood vessels ==> ischemic tissue damage
Pathogenesis - 3 • RBC lasts 17 - 20 days • % of sickled cells correlates to degree of anemia/hemolysis • No correlation b/w sickling and microvascular events • Non-sickled cells with memb damage adhere to the endothelium of the microvessels. • Lowest O2 is in microcirculation
Pathogenesis - spleen • Early childhood spleen enlarged w/ congestion, sickled RBC in splenic cords and sinusoids • Erythrostasis => thrombosis/infarction and tissue hypoxia • Scarring => shrinkage =>autosplenectomy by adolescence • By age 5y 95% have functional asplenia
Immunodeficiency • Impaired splenic function • Defects in alternative complement pathway impairs opsonization of encapsulated bacteria • Increased susceptibility to Salmonella osteomyelitis (bone necrosis) • Pneumococci and Haemophilus influenzae septicemia/meningitis most common causes of death in SCD children
“Crises” • Dactylitis • Splenic sequestration • Aplastic anemia • Acute painful events • Acute chest syndrome • Neurological events
Dactylitis • Common presentation of SCD in children <2 yrs • Swollen, tender hands and feet • Usually bilateral • Ischemia/necrosis of bone • Full resolution expected • Humeral > femoral heads prone to necrosis in older children/adults >50% dev avascular necrosis of femoral head by 35yr
Splenic sequestration • 30% incidence in young children • Rapid, severe worsening of anemia w/ good reticulocytosis • Enlarging, tender spleen • Hypovolemic shock • 15% mortality, 50% recurrent • Often concurrent with viral illnesses • Parents taught to palpate for spleen • Rx: fluid resuscitation. + O2 + transfusion
Aplastic anemia • Young children very sensitive to Parvovirus B19 marrow suppression (68%) • Supraphysiologic oxygen decreases erythropoietin production and decreases retic w/in 2 days • Supportive transfusions until retic return
Acute painful events • Most frequent symptom after age 2 yrs • Vaso-occlusion vs. shunting of blood flow • Rx: hydration, O2 and aggressive narcotic use. Avoid Demerol (incr dependence and possible seizures) • Cause usually not identified • Peripheral/extremities in younger pts and proximal (back, chest, abdomen) in adults
Acute Chest Syndrome • s/s: dyspnea, cough, chest pain • Occasionally: abd pain, fever, tachypnea, infiltrate on CXR, leukocytosis • Major danger is hypoxemia • Affects 30% of SCD w/ Mortality: 10% (higher in adults) • Vaso-occlusion or infection (S. pneum, H. influenzae, Mycoplasma, Chlamydia, Legionella and viruses) • Rx: oxygen, urgent partial exchange transfusion, intubation, ?extracorporeal mb oxygenation, incentive spirometry, +/- bronchodilators • May follow febrile illness in children and painful event in adults (w/ fat emboli)
Acute CNS events • Overt CVA ~ 5% of children w/ SCD • Greatest incidence in 5-10 year olds • Silent infarcts in 17% before age 20 • Correlates with: • seizures • lower painful event rate • incr leukocyte ct >11.8 • incr pocked RBC (>3.5% considered evid of splenic dysfunc) • SEN betaS globin gene haplotype • Cooperative Study of Sickle Cell Disease in USA • Followed patients for 20 years, from 6 months of age • (Pediatrics March 1999)
Stroke • Watershed perfusion areas in most occult CVA • Most strokes are ischemic, rarely hemorrhagic • Anterior vessels of Circle of Willis show progressive narrowing w/ eventual occlusion, collateral vessel dev and occ moyamoya
SICKLE CELL DISEASE IN CHILDREN AND ADOLESCENTS:DIAGNOSIS, GUIDELINES FOR COMPREHENSIVE CARE, AND CARE PATHS AND PROTOCOLS FOR MANAGEMENT OF ACUTE AND CHRONIC COMPLICATIONS* Peter A. Lane, George R. Buchanan, John J. Hutter, Robert F. Austin, Howard A. Britton, Zora R. Rogers, James R. Eckman, Michael R. DeBaun, Winfred C. Wang, Prasad Mathew, Sarah Iden, Michael Recht, Jesse D. Cohen, Ernest Frugé, Leanne Embry, Lewis Hsu, Brigitta U. Mueller, Robert Goldsby, Charles T. Quinn, Marie Mann, and Michele A. Lloyd-Puryear for the Sickle Cell Disease Care Consortium** *Revised at the Annual Meeting of the Sickle Cell Disease Care Consortium, Sedona, AZ, November 10-12, 2001
ACUTE STROKE OR NEUROLOGIC EVENT IN CHILD WITH SICKLE CELL DISEASE DEFINITION: Stroke, defined as an acute, clinically apparent neurological event, occurs in 8-11% of children with Hb SS. Common presenting symptoms and signs include hemiparesis, monoparesis, aphasia or dysphasia, seizures, severe headache, cranial nerve palsy, stupor, and coma. Stroke may occur without warning as an isolated event or may complicate other complications of sickle cell disease such as acute chest syndrome or aplastic crisis. Acute neurologic symptoms or signs require urgent evaluation and treatment. CONSULTS: Hematology Neurology Physical Medicine and Rehabilitation MONITORING: 1. Rapid triage - urgent hematology consultation 2. Hospitalize. Consider ICU admission and/or CR monitor first 24 hr and until stable. 3. Vital signs, neuro checks q 2 hr. 4. Record I & O, daily weight. DIAGNOSTICS: 1. Document duration of acute symptoms, any prior neurologic symptoms or trauma, and results of any previous CNS imaging studies (ie. CT, MRI, MRA, or TCD). 2. Document details of the neurologic exam. 3. Type and crossmatch for transfusion (see Medication/Treatment below). Consider requesting, if available, minor-antigen-matched, sickle-negative, and leukocyte-depleted RBC. 4. CBC, diff, platelet count, and reticulocyte count initially and as clinically indicated (compare with patient's baseline data). 5. RBC minor-antigen phenotype if not previously documented. 6. Consider screening coagulation profile. 7. Blood and urine cultures if febrile. 8. Electrolytes initially and daily until stable. 9. MRI and MRA. If MRI/MRA not immediately available, CT without contrast to exclude intracranial hemorrhage with MRI/MRA later when available. Initiation of transfusion therapy should not be delayed by arrangements for imaging studies. 10. Consider CSF culture if febrile and no contraindication present. FLUIDS, GENERAL: 1. IV + PO@ 1 x maintenance MEDICATION/TREATMENT: 1. Partial exchange transfusion or erythrocytapheresis to Hb 10 gm/dl and Hb S (patient's RBC) 30% (may require transfusion medicine consult for erythrocytapheresis). Remove femoral or central venous catheter as soon as possible after exchange transfusion to reduce risk of thrombosis. 2. Simple transfusion with RBC to Hb approximately 10 gm/dl may be considered as an alternative to partial exchange transfusion for stable patients with Hb <6-7 gm/dl (do not transfuse acutely to Hb >10 gm/dl, Hct >30%). 3. Rx seizures if present. 4. Rx increased intracranial pressure if present. 5. 02 by nasal cannula or face mask if needed to keep pulse ox 92% or patient's baseline, if >92%. The etiology of a new or increasing supplemental 02 requirement should be investigated. 6. Consider hemoglobin electrophoresis after partial exchange transfusion or at discharge. 7. Cefotaxime or cefuroxime 50 mg/kg IV q 8 h if febrile. Substitute clindamycin 10 mg/kg IV q 6 hr for known or suspected cephalosporin allergy. Strongly consider adding vancomycin 10-15 mg/kg IV q 8 hr for severe febrile illness or for proven or suspected CNS infection. 8. If applicable, continue prophylactic penicillin. Prophylactic penicillin should be discontinued while patient is receiving broad-spectrum antibiotics. 9. See other Clinical Care Paths for pain, acute chest syndrome, acute splenic sequestration, aplastic crisis, priapism, if present. DISCHARGE CRITERIA: 1. Clinically and neurologically stable 24 hr. after transfusions. 2. Afebrile 24 hr. with negative cultures for 24-48 hr. if applicable. 3. Taking adequate oral fluids and able to take oral medication if applicable. 4. Hematology, rehabilitation, and physical therapy follow-up organized. 5. Initiate chronic transfusion program (see p. 27).
ACUTE STROKE OR NEUROLOGIC EVENT IN CHILD WITH SCD DEFINITION: Stroke, defined as an acute, clinically apparent neurological event, occurs in 8-11% of children with Hb SS. Common presenting symptoms and signs include hemiparesis, monoparesis, aphasia or dysphasia, seizures, severe headache, cranial nerve palsy, stupor, and coma. Stroke may occur without warning as an isolated event or may complicate other complications of sickle cell disease such as acute chest syndrome or aplastic crisis. Acute neurologic symptoms or signs require urgent evaluation and treatment.
DIAGNOSTICS: 1. Document duration of acute symptoms, any prior neurologic symptoms or trauma, and results of any previous CNS imaging studies (ie. CT, MRI, MRA, or TCD). 2. Document details of the neurologic exam. 3. Type and crossmatch for transfusion (see Medication/Treatment below). Consider requesting, if available, minor-antigen-matched, sickle-negative, and leukocyte-depleted RBC. 4. CBC, diff, platelet count, and reticulocyte count initially and as clinically indicated (compare with patient's baseline data). 5. RBC minor-antigen phenotype if not previously documented. 6. Consider screening coagulation profile. 7. Blood and urine cultures if febrile. 8. Electrolytes initially and daily until stable. 9. MRI and MRA. If MRI/MRA not immediately available, CT without contrast to exclude intracranial hemorrhage with MRI/MRA later when available. Initiation of transfusion therapy should not be delayed by arrangements for imaging studies. 10. Consider CSF culture if febrile and no contraindication present
FLUIDS, GENERAL: 1. IV + PO@ 1 x maintenance
MONITORING: 1. Rapid triage - urgent hematology consultation 2. Hospitalize. Consider ICU admission and/or CR monitor first 24 hr and until stable. 3. Vital signs, neuro checks q 2 hr. 4. Record I & O, daily weight.
MEDICATION/TREATMENT: 1.Partial exchange transfusion or erythrocytapheresis to Hb 10 gm/dl and Hb S (patient's RBC) 30% (may require transfusion medicine consult for erythrocytapheresis). Remove femoral or central venous catheter as soon as possible after exchange transfusion to reduce risk of thrombosis. 2. Simple transfusion with RBC to Hb approximately 10 gm/dl may be considered as an alternative to partial exchange transfusion for stable patients with Hb <6-7 gm/dl (do not transfuse acutely to Hb >10 gm/dl, Hct >30%). 3. Rx seizures if present. 4. Rx increased intracranial pressure if present.
MEDICATION/TREATMENT 5. 02 by nasal cannula or face mask if needed to keep pulse ox 92% or patient's baseline, if >92%. The etiology of a new or increasing supplemental 02 requirement should be investigated. 6. Consider hemoglobin electrophoresis after partial exchange transfusion or at discharge. 7. Cefotaxime or cefuroxime 50 mg/kg IV q 8 h if febrile. Substitute clindamycin 10 mg/kg IV q 6 hr for known or suspected cephalosporin allergy. Strongly consider adding vancomycin 10-15 mg/kg IV q 8 hr for severe febrile illness or for proven or suspected CNS infection. 8. If applicable, continue prophylactic penicillin. Prophylactic penicillin should be discontinued while patient is receiving broad-spectrum antibiotics. 9. See other Clinical Care Paths for pain, acute chest syndrome, acute splenic sequestration, aplastic crisis, priapism, if present.
DISCHARGE CRITERIA: 1. Clinically and neurologically stable 24 hr. after transfusions. 2. Afebrile 24 hr. with negative cultures for 24-48 hr. if applicable. 3. Taking adequate oral fluids and able to take oral medication if applicable. 4. Hematology, rehabilitation, and physical therapy follow-up organized. 5. Initiate chronic transfusion program (see p. 27).
CVA treatment • Risk of recurrence is 70-90% and may be fatal • Chronic monthly transfusion regimen to maintain HbS <30% • Reduces recurrence by 90% • Continue until 18 years old • Transcranial Doppler ultrasound to assess velocity of flow in circle of Willis every year to screen children • Not readily available for children here • MRI/MRA • If velocity >200 cm/sec, stroke risk is 10% per year • start monthly transfusion therapy • Reduces risk of stroke by 90% • Recommendation from the Stroke Prevention in Sickle Cell Anemia Trial (STOP)