Download

1 / 26
260 likes | 560 Views
The International Tomato Sequencing Project: The first Cornerstone of the SOL Project. Lukas Mueller on behalf of International SOL Tomato Sequencing Project. Overview. Aims Why sequence the tomato genome? How to sequence the tomato genome? Who is sequencing the tomato genome?

E N D
The International Tomato Sequencing Project: The first Cornerstone of the SOL Project Lukas Mueller on behalf of International SOL Tomato Sequencing Project
Overview • Aims • Why sequence the tomato genome? • How to sequence the tomato genome? • Who is sequencing the tomato genome? • Resources for Sequencing the Tomato Genome • Genetic Map • BAC libraries • Overgo mapping • BAC End Sequences • Minimal Tiling Path • Bioinformatics • Summary
Ramesh Sharma Jiten Khurana Akhilesh Tyagi Doil Choi Byung Dong Kim Mingsheng Chen Zhukuan Cheng Chuanyou Li Hongqing Ling Yongbiao Xue Antonio Granell Miguel A. Botella Giovanni Giuliano Luigi Fruciante Daisuke Shibata Satoshi Tabata Graham Seymour Gerard Bishop Steven D. Tanksley Jim J. Giovannoni Stephen Stack, Joyce van Eck Mondher Bouzayen Mathilde Causse Willem Stiekema P. Lindhout Taco Jesse Rene Klein Lankhorst
Aims • Provide a high quality reference sequence for the Solanaceae genomes • Using mapping of other Solanaceae sequences onto the tomato sequence, and comparative genetic maps to derive “virtual” genomes for other Solanaceae • Prerequisite for studying natural diversity and linking genotype to phenotype • Build a Solanaceae bioinformatics platform to integrate, analyze and distribute the information
WHY SOLANACEAE? Solanaceae is part of unique clade of flowering plants. Genome research in Solanaceae will provide a reference anchor and enable comparative genomics and systematic throughout this clade Solanaceae Rubiaceae (coffee) asterid I Compositeae (sunflower, safflower, lettuce) asterid II asterid III asterid IV asterid V Leguminosae (soybean, Medicago rosid I Rosaceae (apple, peach, cherry); Salicaceae (poplar) Malvaceae (cotton) ; Sterculiaceae (cocoa) rosid II Arabidopsis ; Rutaceae (citrus) Brassicaceae rosid III Chenopodiaceae (sugarbeet, spinach) caryophyllids hamamelid I hamamelid II ranunculids paleoherb II Magnoliales Rice Gramineae (maize, wheat) ; Musaceae (banana) monocots Liliaceae (onion) Laurales
Why sequence tomato? • Tomato is the most intensively researched Solanaceae genome encoding approx. 35,000 genes euchromatic regions corresponding to less than a 25% of the total DNA in the tomato nucleus (220~250 Mb). • Tomato provides the smallest diploid genome for which homozygous inbreds are available. • Its sequence will facilitate positional cloning in tomato and other Solanaceae genomes (via synteny maps).
How to sequence the tomato genome? • Whole Genome Shotgun • Advantages: Fast, cheaper, ok with reference genome • Disadvantages: Unordered contigs • Methylation Filtering (Tobacco) • Advantages: Selects for expressed genome, cheaper • Disadvantages: unordered contigs • Tiling Path (Arabidopsis, Drosophila, Rice) • Advantages: Sequence and gene order; select gene rich regions; easy to divide work • Disadvantages: Relatively expensive, time consuming • ORDER IMPORTANT FOR COMPARING GENOMES
telomere euchromatin telomere structure pericentric heterochromatin 162 bp sub-telomeric repeat centromere pericentric heterochromatin 7 bp telomeric repeat euchromatin Tomato Genome Structure • 12 chromosomes • 950MB of total DNA • 220MB contiguous, gene rich euchromatin • Sequence only gene-rich euchromatin (>90% all genes) • Tiling path method preferred • Drosophila used and Medicago is using similar strategy
BAC libraries • All libraries derived from Solanum lycopersicum Heinz 1706. • HindIII library (Rod Wing, Clemson U) • ~120,000 clones, 120kB average size • ~15x coverage • FPC contigged • Overgo analysis • 75,000 clones BAC end sequenced • MboI library • 50,000 clones, 140kb average size • Will be BAC end sequenced • EcoRI library (being prepared) • Will be BAC end sequenced
F2-2000 Genetic Map • Parents: • Solanum lycopersicum x Solanum pennellii • Mapping population of 80 F2 individuals • # Markers: 1579 • Total cM: 1453 • Density: 1 marker/0.92cM • SGN http://sgn.cornell.edu/ Marker-Types: rflp 345 ssr 149 tm 43 p-mrkr 39 cos 576 est-by-clone 265 unknown 8 caps 21 cosii 98 kfg 35 Total 1579
Tying the Genetic Map to the Physical Map: Overgos • Overgos are “overlapping oligos”, short, very hot probes, developed from genetic markers of the F2-2000 map • Overgos are organized in 96 well plates, analyses are carried out with row and column pools • Pools are hybridized to BAC filters, raw pool results are deconvoluted • A total of 1536 overgos developed (16 plates) • Analyses of all plates is complete
Overgo Anchoring Results Anchors: • 652 anchor markers are involved in plausible non-conflicted associations with BACs. • 4857 good marker--BAC associations FPC contigs: • 1880 BACs in 705 plausible contigs • 2166 BAC singletons • 652 seed BACs ==> 1/3 of euchromatic genome sequence
92 165 1.8 79 143 1.8 67 171 2.6 62 137 2.2 40 119 3.0 63 101 1.6 51 112 2.2 34 87 2.6 40 116 2.9 41 87 2.1 43 103 2.4 39 120 3.1 # anchors cM chr length cM per anchor Distribution of Anchor Markers on Chromosomes + 1000 markers from Keygene AFLP map
Verification of overgo mappings • Fluorescence In-Situe Hybridization (FISH) • BAC probe on pachytene chromosomes • IL lines (Zamir lab) • Map BACs to IL lines • CAPS assays
Summary of FISH verification • Song-Bin Cheng, Hans de Jong (Holland, chromosome 6): • 9 BACs analyzed • 8 mapped to chromsome 8 in right order • 1 BAC gave signals on centromere of chromosome 1 • Sangheob Lee, Doil Choi (Korea, chromosome 2): • 27 BACs analyzed with FISH • 25 confirmed to specific location, same order as F2-2000 map • 2 match to other chromosomes • Chuanyou Li (China, chromosome 3) • >30 BACs being analyzed • Steven Stack (USA): • Telomere and heterochromatic boundary determination • FISH service for countries without FISH capability
BAC end sequences • Total of 400,000 reads (200,000 BACs from both ends) selected from the 3 BAC libraries • Batch of 75,000 BACs in process (HindIII library) • ~45,000 BAC end sequences already obtained (ftp://ftp.sgn.cornell.edu/tomato_genome/) • Average read length 655bp • Annotation in progress • SeqWright Inc, Houston, TX • SeqWright is sponsoring a happy hour after this session.
C A B anchored bacs Obtaining the Tiling Path overgos genetic map “seed BAC” “seed BAC”
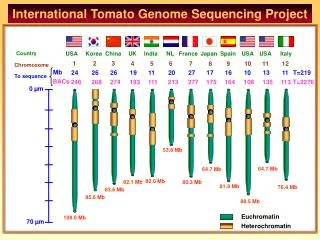
US Korea China UK India NL France Japan Spain US US Italy BACs finished: in process: 4 14 16 20 9 5 5 Overview: sgn.cornell.edu -> About -> tomato sequencing
Building a Bioinformatics Platform for the Solanaceae • Project-wide standards for quality, gene naming, annotation (http://sgn.cornell.edu/solanaceae-project/) • Create a unified web presence for the entire project • Develop distributed model for annotation, web presentation, involving different centers in SOL countries • All data and programs developed in the project are shared in an open source format • Integrate all data into the SOL bioinformatics platform, facilitating a systems approach to explore diversity and adaptation and the complex interactions that occur on all levels of biological organization
CAS Genome India SGN Agronanotech Kazusa VIB Ghent
Annotation Phases • First pass annotations of sequences and gene models on BAC basis, available immediately • BAC based, common, distributed platform, stable BAC-based identifiers • Chromosome based, stable identifiers
Summary • Sequencing of tomato is under way by a consortium of 10 countries • High quality, ordered sequence using BAC tiling path • BAC ends available, overgo results verified by FISH analyses • Sequence will be tied to other Solanaceae and closely related species (coffee and beyond) • Provide a foundation for shared biology for this economically important clade of plants
SOL community Tomato Sequencing Project Funding National Science Foundation Other National Funding Sources Keygene NV Seqwright Inc. (Happy Hour) Colleagues Steven Tanksley, Jim Giovannoni, Joyce van Eck , Steven Stack SGN: Teri Solow, Beth Skwarecky, Nick Taylor, Robert Buels, John Binns, Chenwei Lin Acknowledgments