Download
1 / 1
70 likes | 609 Views
Calculate simulated conversion. Calculate Objective Function. High Temperature Water Gas Shift Reaction over Iron Oxide Catalysts Rainee VanNatter, Carl RF Lund. Load Experiment. Introduction. Calculate Rate Constants. CO + H 2 O ↔ H 2 + CO 2.

E N D
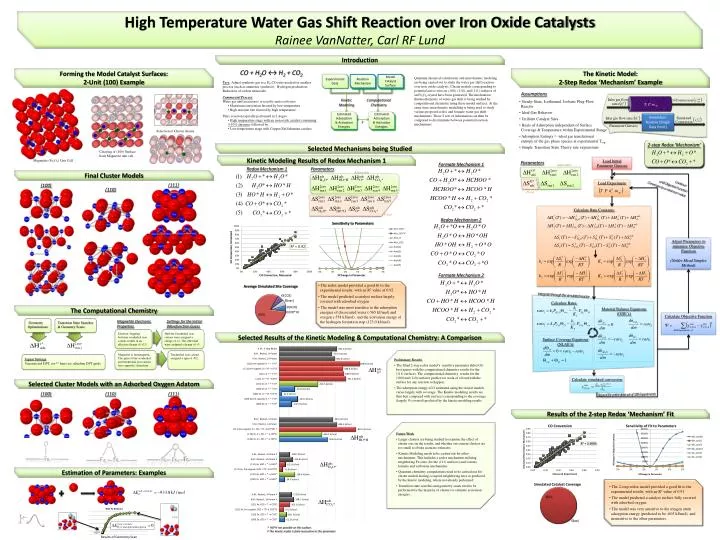
Calculate simulated conversion Calculate Objective Function High Temperature Water Gas Shift Reaction over Iron Oxide Catalysts Rainee VanNatter, Carl RF Lund Load Experiment Introduction Calculate Rate Constants CO + H2O ↔ H2 + CO2 Forming the Model Catalyst Surfaces: 2-Unit (100) Example The Kinetic Model:2-Step Redox ‘Mechanism’ Example Experimental Data Reaction Mechanism Model Catalyst Surface Quantum chemical calculations and microkineticmodeling are being carried out to study the water gas shift reaction over iron oxide catalysts. Cluster models corresponding to potential active sites on (100), (110), and (111) surfaces of an Fe3O4 crystal have been generated. The mechanistic thermochemistry of water-gas shift is being studied by computational chemistry using these model surfaces. At the same time, microkinetic modelling is being used to study various proposed redox and formate water gas shift mechanisms. These 2 sets of information can then be compared to discriminate between potential reaction mechanisms. • Uses: Adjust synthesis gas to a H2:CO ratio needed for another process (such as ammonia synthesis); Hydrogen production; Reduction of carbon monoxide. • Commercial Process:Water gas shift reaction is reversible and exothermic. • Maximum conversion favored by low temperature • High reaction rate favored by high temperature. • Thus, reaction typically performed in 2 stages: • • High temperature stage with an iron oxide catalyst containing 5-10% chromia; followed by • • Low temperature stage with Copper/ZnO/alumina catalyst. Assumptions Kinetic Modeling Computational Chemistry • Steady-State, Isothermal, Isobaric Plug-Flow Reactor • Ideal Gas Behavior • Uniform Catalyst Sites • Heats of Adsorption independent of Surface Coverage & Temperature within Experimental Range • Adsorption Entropy ≈ -ideal gas translational entropy of the gas phase species at experimental Tavg. • Simple Transition State Theory rate expressions Estimated Adsorption & Activation Energies Estimated Adsorption & Activation Energies ? Parameters Parameters Adjustable Adjustable Selection of Cluster Atoms Selected Mechanisms being Studied Cleaving of (100) Surface from Magnetite unit cell Fixed Kinetic Modeling Results of Redox Mechanism 1 Magnetite (Fe3O4) Unit Cell Formate Mechanism 1 Calculate Rates Fixed Redox Mechanism 1 2-step Redox ‘Mechanism’ Final Cluster Models (111) (100) (110) Continue . until Objective Function Converges to a minimum value Load Initial Parameter Guesses Material Balance Equations(ODE’s) Surface Coverage Equations(NLAE’s) Redox Mechanism 2 T, P, mcat Adjust Parameters to minimize Objective Function(Nelder Mead Simplex Method) Simulation Routine (Single Data Point) Formate Mechanism 2 • The redox model provided a good fit to the experimental results, with an R2 value of 0.92 • The model predicted a catalyst surface largely covered with adsorbed oxygen • The model was most sensitive to the adsorption energies of dissociated water (-565 kJ/mol) and oxygen (-558 kJ/mol), and the activation energy of the hydrogen formation step (123.0 kJ/mol). The Computational Chemistry Magnetite Electronic Properties Settings for the Initial Wavefunction Guess Geometry Optimizations Transition State Searches & Geometry Scans Integrate through the simulated reactor Electron ‘hopping’ between octahedral iron cations results in an effective charge of +2.5 Half the Octahedral iron cations were assigned a charge of +2. The other half were assigned a charge of +3. Selected Results of the Kinetic Modeling & Computational Chemistry: A Comparison Magnetite is ferrimagnetic. The spins of the octahedral and tetrahedral iron cations have opposite ‘directions’. Tetrahedral iron cations assigned a spin of -5/2. Preliminary Results • The fitted 2-step redox model’s sensitive parameter (hloc(O)) best agrees with the computational chemistry results for the (111) surfaces. The computational chemistry results for the (100) and (110) surfaces predict too weak of a bond with the surface for any reaction to happen. • The adsorption energy of O estimated using the cluster models varies largely with coverage. The Kinetic modeling results are thus best compared with surfaces corresponding to the coverage (largely O-covered) predicted by the kinetic modeling results. Jaguar Settings Unrestricted DFT, tzv** basis set, ultrafine DFT grids Selected Cluster Models with an Adsorbed Oxygen Adatom (100) (110) (111) Results of the 2-step Redox ‘Mechanism’ Fit Repeat for entire data set of 189 Experiments Future Work • Larger clusters are being studied to examine the effect of cluster size on the results, and whether our current clusters are too small to obtain accurate estimates. • Kinetic Modeling needs to be carried out for other mechanisms. This includes a redox mechanism utilizing neighboring Fe-sites (for the (111) surfaces) and various formate and carbonate mechanisms. • Quantum chemistry computations need to be carried out for cluster models having occupied neighboring sites as predicted by the kinetic modeling, where not already performed. • Transition state searches and geometry scans need to be performed for the majority of clusters to estimate activation energies. Estimation of Parameters: Examples • The 2-step redox model provided a good fit to the experimental results, with an R2 value of 0.91 • The model predicted a catalyst surface fully covered with adsorbed oxygen • The model was very sensitive to the oxygen atom adsorption energy (predicted to be -605 kJ/mol), and insensitive to the other parameters. † HO*H not possible on this surface. ‡ This kinetic model is fairly insensitive to this parameter Results of Geometry Scan