Download

1 / 18
180 likes | 474 Views
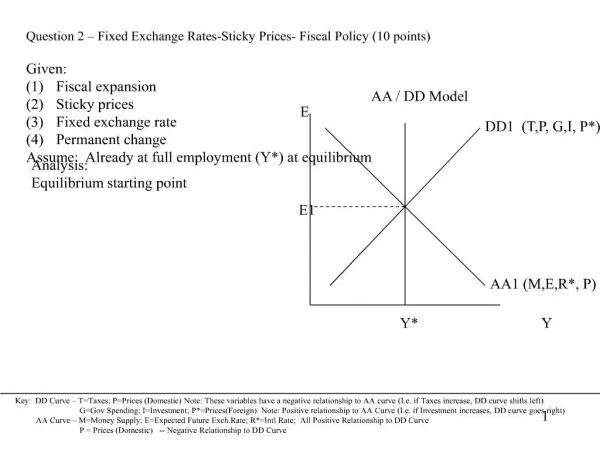
FDA AA - Title VIII. Clinical Trial Databases Friday, 30 November 2007 MBC Sarah Doyle Larson Program Manager, Biomedical Regulatory Affairs Compliance. Agenda. Title VIII Overview Timelines Clinical Trial Registry Clinical Trial Results Database Other Provisions

E N D
FDA AA - Title VIII Clinical Trial Databases Friday, 30 November 2007 MBC Sarah Doyle Larson Program Manager, Biomedical Regulatory Affairs Compliance
Agenda • Title VIII Overview • Timelines • Clinical Trial Registry • Clinical Trial Results Database • Other Provisions • Other Non-FDA Influences to Consider • State of Maine • WHO • ICMJE • Trade Associations • International Regulations and Standards
What is Title VIII? • Definitions • Registry = Protocol parameters for ongoing trials • Results Database = Results data from completed trials • Title VIII Mandates the expansion of www.ClinicalTrials.gov to form a Clinical Trials Registry and Results Database • Broader scope of trials that must be registered • Increases the amount of data elements required in each posting • Requires submission of clinical trial results data • Penalties for noncompliance • Several “other” requirements • Implementation is a “phased” approach • Timelines for complying range from Dec. 26, 2007 to Sep. 27, 2010
FDA/NIH Implementation Timeline1 3 years (9/27/2010) Expansion of registry and results database by rulemaking • 90 Days (12/26/07) • Linking to FDA and NIH information • Expanded registry data elements • Expanded registry scope • 18 months (3/27/09) • Public meeting to discuss expanded registry and results database • Adverse events (if by rulemaking) CongressPassesLaw • 1 Year (9/27/08) • Basic results reporting • Expanded registry scope 2 Years (9/27/09) Adverse events (if by default) 2010 Sept 27 2007 2008 2009 1Timeline provided by Terry Toigo, Director, FDA Office of Special Health Issues, 27 Nov 2007
Expanded Registry Data Bank – 26 Dec 2007 • Scope of trials expanded beyond FDAMA §113 • Drug/Biologics: All controlled clinical investigations, other than Ph. 1 • Devices: All controlled trials with health outcomes; excludes small feasibility studies; includes pediatric postmarket surveillance • Additional Information Required: • Some previously optional fields are now mandatory • New data elements • Links to “results” information to be provided by FDA / NIH • Maintaining Postings: • Updates – at least once/every 12 months • Recruitment Status – w/in 30 days of change • Penalties for Noncompliance and Public Notices • NIH Implementation & Compliance: 90 Days (26 Dec 2007)
Expanded Registry Data Bank – When to Register? • Newly initiated applicable trials: • register by later of: 26 Dec 2007 -OR- 21 days > 1st pt enrolled • “Ongoing” applicable trials as of 27 Sep 2007 that are not “completed” by 26 Dec 2007 … • if SLT = “yes”, update posting to include all req’d fields by: 26 Dec 2007 • if SLT = “no”, update posting to include all req’d fields by:27 Sep 2008 • Notes regarding NIH “posting” dates: • Drugs OR approved/cleared Devices • w/in 30 days of receipt • Devices not previously approved/cleared • “held” for release until approved; then released w/in 30 days of approval
Clinical Trial Results Data Bank • Results Links • Basic Results • Expanded Results Data Bank
Results Data Bank “Links” – 26 Dec 2007 • Scope: • Approved Drug, Biologics, Devices • CTs that form basis for efficacy claim or CTs conducted > approval • FDA Information to be posted (Links): • Advisory Committee Meetings • FDA assessment of results for pediatric trials (Sec 505A/B) • FDA public health advisories • FDA drug “action package” for approval documents • FDA device PMA or 510(k) summary of safety/efficacy • NIH Information to be posted (Links): • Medline citations • Label • Implementation Timeline: “Beginning” 26 Dec 2007 • Responsibility of FDA/NIH • For applicable trials ongoing as of 27 Sep 2007, link 30 days > approval and w/in 30 days of results becoming public • For other trials posted prior to 27 Sep 2007: as available
Results Data Bank “Basic Results”– 27 Sep 2008 • Scope: • Approved Drugs, Biologics, Devices • Includes applicable trials completed per protocol & terminated trials • Information to be posted: • Demographics & baseline characteristics of patient sample • Primary & Secondary Outcomes • Point of Contact • Certain Agreements • Implementation Timeline: 27 Sep 2008 • In general, submit w/in 12 mos of completion date (estimated or actual) • Completion Date = LPLV • Delayed submission w/ certification: • Initial approval: Submit results w/in 30 days of initial approval/clearance • New use: Submit results w/in 30 days of • Approval/clearance of new use • Issuance of letter by FDA (e.g. complete response, not approving/clearing, not approvable) • Withdrawal w/out resubmission after 210 days • Limit if no action: 2 years
Results Data Bank “Expanded Data Bank” 27 Sep 2010 • Public meeting to be held by 27 Mar 2009 • Rulemaking for AE Reporting by 27 Mar 2009 (or default provision will apply on 27 Sep 2009) • Rulemaking for Expansion of Data Bank by 27 Sep 2010: • Scope: • Trials w/ completion date on or > Sept 1997 for approved products • May include unapproved products (whether approval was sought or not) • Information and format to be posted: • Non-technical & technical summaries of results (non promotional) • Protocol or other info needed to “evaluate” completeness of results • “Other categories as determined to be appropriate” by FDA • Results Submittal Timeline – extend from 12 mos to 18 mos? • How to address trials where results were posted < data bank “Expansion”? • Implement QC Procedures • Consider WHO Data Set
Other Provisions in Title VIII • Certification w/ all submission - 27 Dec 2007 • State Preemption – 27 Dec 2010 • Modify ICF • Public Notice of Noncompliance; Penalties • Guidance to be issued re: how requirements apply to a pediatric postmarket surveillance • Voluntary submissions acceptable • QC Pilot Program • confirm completeness, accuracy, and ensure non-promotional • Consult w/ experts on risk communication • Waiver process for “extraordinary circumstances” when protecting public health or national security
Other Considerations • US States: • Maine - passed March 2007; FDAAA preemption effective 27 Sep 2010 • Other State activity in 2007 • WHO: • Policy: registration & results of ALL human subject trials • WHO Primary Registers • WHO data elements • ICMJE – • WHO data elements • Require prospective registration of “any research project that prospectively assigns human subjects to intervention or comparison groups to study the cause-and-effect relationship between a medical intervention and a health outcome” • As of July 2008, will no longer exclude Phase I • Ingelfinger Rule / Post results abstract <300 words if pub is pending • Trade Associations • PhRMA - joint position statement Jan 2005 • BIO - Principles on Clinical Trials • Advamed
Varying International Standards • Mandatory • Israel (clinicaltrials.gov) • Italy (local language; country specific EUDRACT posting) • South Africa (country specific) • Taiwan • Proposed • France (local language; requirements similar to Italy) • Voluntary • Australia (country specific website) • China (country specific website) • Germany • India (country specific website) • Japan (country specific website) • Netherlands • Spain • UK (country specific website) • Local IRB/EC requirements
Summary • New FDAAA requirements begin 26 Dec 2007 and will continue through 27 Sep 2010 • FDA / NIH is still in the process of implementing … Stay tuned! http://www.clinicaltrials.gov http://prsinfo.clinicaltrials.gov • In some cases, the NIH’s timeline for implementation allows little time to update internal SOPs and/or to complete tasks as required. • Rulemaking! • Lack of harmonization between FDAAA and other “influences”