Download

1 / 38
460 likes | 912 Views
Meta-Genomics by 16S. Mark Reimers, Moscow Genomic Data Analysis 2012. Outline. What is Metagenomics ? biological questions and statistical issues What’s there? Identification issues How have things changed? Characterizing differences in microbial communities. What is Meta-genomics?.

E N D
Meta-Genomics by 16S Mark Reimers, Moscow Genomic Data Analysis 2012
Outline • What is Metagenomics? • biological questions and statistical issues • What’s there? Identification issues • How have things changed? Characterizing differences in microbial communities
What is Meta-genomics? • Unbiased survey of microbial communities by sequencing and identifying DNA • Most bacteria cannot be easily cultured • Sequencing random fragments of DNA from all microbial denizens of a community (and traces of a few eukaryotes) • Term is alsoused for surveys of microbial diversity based on sequencing all 16SrRNA genes present
Kinds of Questions • What species are there? • Most microbial species are not known • What microbes and ecosystems associated with specific conditions? • Pollution or toxicity • Infectious disease • Non-infectious disease? • What drives the dynamics of the community?
Environmental Meta-Genomics • Craig Venter 2003 Ocean Sampling Expedition – Sargasso Sea and other regions • 2006 - 454 sequencing of microbial samples • Questions: Diversity, ecosystem dynamics, interactions with plants, animals and protists, markers for pollution, community metabolism
Human Microbiome Project • Catalog diversity of all microbes resident in and on human body, and their relationship to disease
Diversity of External Microbes • Considerable diversity in different skin locations • Surprising number of anaerobes (Bacteroides)
Meta-Genomic Shotgun Assay • Dissolve cell walls and extract DNA from mixed sample of micro-organisms • Add random primers to extracted DNA • Amplify by PCR • Sequence • Need very long reads (> 100 bases) to identify organisms or gene types • BLAST against database of genomes of presumably related organisms • COG – Clusters of orthologous groups • Count hits against each species and each type of gene • (Assemble genomes of dominant organisms)
Advantages and Disadvantages of Microbe Shotgun Sequencing • Can sample metabolic or infectious potential of microbes • Same species may have different variants with very different properties – e.g. E. coli or S. aureus • Better able to address questions of metabolic activity if organisms are uncharacterized by lab work (most organisms) • Tedious and expensive
Biases in Shotgun Metagenomics • Extraction bias: some organisms have differently packaged DNA or tougher cell walls (more an issue with eukaryotes) • GC bias: sequences with higher GC content are more amplified by PCR • Genome size: genes from organisms with bigger genomes will show up more often • Length distribution of different sequences varies • Many earlier 454 could not be BLASTed against COG
Identifying Taxa by 16S • 16S ribosomal RNA is the RNA component of the small sub-unit of the bacterial ribosome (S = Svedberg sedimentation rate) • Highly conserved within species • Typical within-species identity 99% • Typical within-genus between-species identity ~90%-95%
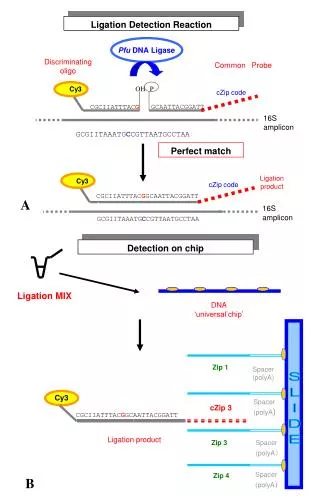
16S Metagenomic Assay • Extract DNA from sample • PCR amplify with ‘universal primer’ for one highly conserved section of rRNA gene in order to capture some variable regions: • V1-V3;V3-V5; V7-V9 • Sequence amplified fragments • Compare reads to database of known variable regions to identify reads
Advantages and Disadvantages of Microbe 16S Sequencing • Can identify many more microbes for the same sequencing effort • So-called ‘universal primers’ do not amplify well 16S rRNA genes from some organisms • May not be able to assign all taxa from a sample at the same level! • Modest differences in sequencing artifacts can bias identifiability of fragments between samples • Some ambiguity about strains
Analytic Issues for 16S Metagenomics • How to identify different microbes • How to characterize differences • Representation biases
16S Assay Identification Issues • How to decide whether similar variants are from same or different species? • Ad-hoc rule: 98% identity -> same species • Some evidence that even 99% may be too low • How to distinguish errors in sequencing from novel microbes? • Use knowledge of typical sequence biases • e.g. 454 often cannot resolve 4 or 5 repeats • AAAA or AAAAA?
Identifying Microbes by 16S • Databases: 2007 MEGAN; 2009 RDP • Not all species may be represented
Identifying Unknown Organisms - OTU • Can cluster sequences within each sample to construct Operational Taxonomic Units (OTU) • These may coincide with species (if lucky) • Then identify clusters in further samples • Problems • On data with known species, these methods frequently seriously over- or under-cluster genera or species
Correspondence of Metagenomic and 16S Assays • Relative abundance of common organisms is well correlated between assays • Correspondence of variations of organisms across samples has not been tested
Distribution of Abundances – Power Law Again Collapsing across the entire dataset, the number of times each sequence (or OTU at 100%) is observed is approximately linear in log-log space across six orders of magnitude The rare sequences likely reflect a mix of sequencing error and true rare taxa. Using other sources of information (Sanger 16S amplicon sequencing, 454 and Illumina whole-genome metagenome), analysis groups within HMP are trying to discriminate true taxa from noise.
Data Analysis Issues – 16S Assay • How to deal with unrecognized reads • Replicability and stability of measures • How to estimate true abundance given fragment count • How to compensate for changes in sequencing biases • How to adjust for sequencing depth • How to test if a species abundance differs meaningfully between groups
Operational Taxonomic Units for Unrecognized Sequences • Many sequences cannot be classified • However many clusters of 99% identity can be recognized among them • How do we best cluster unknown reads? • This problem will gradually attenuate
Different Levels of Identification • Some reads can be classified as to species, others only to genus, others only to family or even order • Many unknown species within a genus • Sequencing errors may prevent precise identification • Level of sequencing errors varies between samples • Confidence levels in classification may be useful • How to do statistical testing?
Microbes are Represented Unevenly by 16S Metagenomics Proportions of 22 identified microbes in samples prepared with equal numbers
Representation Biases Vary Systematically PCA plot showing VCU mock samples, colored by month
Correlation of Proportion among Assigned Reads with Proportion of All Reads Assigned at Finest Level • Lactobacillus • 60% • Gardnerella • 34%
16S – Representation Biases • rRNA from different organisms seems to be represented very differently • Proportions of sequences not representative Variation in read lengths for different organisms Proportions of 22 identified microbes in samples prepared with equal numbers
Representation Biases Vary from Sample to Sample Distributions of read lengths colored by genus in 12 samples • Typically a minimum read length cutoff is imposed and shorter reads are dropped • Changes in read abundance for some microbes may not reflect real changes
Characterizing Microbial Communities • Current practice is tocluster microbial ecosystems to show relationships • Euclidean, correlation or Manhattan metrics are inapplicable with high dispersion • No consensus on clustering metric • Square or cube root transform may work • log emphasizes small values too much
Testing for Differences in Microbes • Most people test for differences between community distributions • Dirichlet distribution on sets of proportions is a way of modeling distribution of distributions • Many biologists are interested in which microbes are differently represented between samples • Multiple comparisons problem again • Skewed distribution makes testing difficult
Significance Testing for Microbe Data • Distributions are highly skewed: more zeros than would be expected from any standard distribution with same mean
Approaches to Skewed Distributions • Continuous approach – model proportions • Use mixture model with some probability of 0, then some distribution, often exponential, of counts • Need to estimate both parameters for both groups • Discrete approach – model counts • Fit proportions - adjust for total number of reads • Too many zeros to fit Negative Binomial
Zero-Inflated Negative Binomial Model From T. Guennel, PhD Thesis, 2012
16S Metagenomic Analysis – a Summary • Identification issues will be resolved over the next few years for common ecosystems • Many data analysis issues are still unresolved • Plausible models for testing community structure differences exist • Almost any model for testing single microbes (or sets) will require a mixture • Unclear how to do significance testing