Download

1 / 35
350 likes | 523 Views
IND Process and General Responsibilities under IND August 27, 2010 Kate Marusina, PhD, MBA Pav Aujla, MS, CCRP, RAC Primo N. Lara, Jr. MD. A Translational Innovation Forum. Definition of Drug and other helpful definitions 21 CFR 312 What is Regulatory Sponsor? Overview of IND process

E N D
IND Process and General Responsibilities under INDAugust 27, 2010Kate Marusina, PhD, MBAPav Aujla, MS, CCRP, RACPrimo N. Lara, Jr. MD A Translational Innovation Forum
Definition of Drug and other helpful definitions 21 CFR 312 What is Regulatory Sponsor? Overview of IND process General Responsibilities under IND Agenda
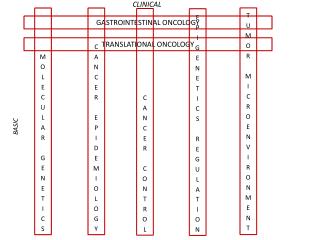
University of California Davis Cancer Center – Clinical Trials Support Unit (CTSU) Best Practices for: IND Exemptions: Consultative Process Protocol Development Protocol Initiation Meeting Post Study on http://www.clinicaltrials.gov Agenda
Good Laboratory Practices (GLPs) for non-clinical studies 21 CFR 58 Good Manufacturing Practices 21 CFR 210, 211 Comprehensive regulations to assure the identity, strength and purity Key Compliance Documents
Good Clinical Practices (GCPs) Protection of Human Subjects 21 CFR 50 Financial Disclosure of Investigators 21 CFR 54 Institutional Review Boards 21 CFR 56 Sponsor, Monitor and Investigator Obligations 21 CFR 312, Subpart D Key Compliance Documents
“ Any drug (or biologic) that is not generally recognized as safe and effective for use under the conditions prescribed, recommended or suggested in the labeling thereof” - Section 201 FD&C Act What is a New Drug?
FD&C Act prohibits shipment of any drug across the state lines without an approved NDA Investigational New Drug (IND) Application provides an exemption to allow shipment of the drug for clinical testing What is a New Drug?
New Molecular entity A pharmacologically active moiety which is not been studied or used clinically in man or is not approved for such use Drug Substance (API) Pharmacologically active Subsequently formulated with excipients to produce the drug product Drug Product Finished product in certain dosage form (capsule, tablet, injection) Some Useful Definitions
Commercial Research (non-commercial), also called an Investigator IND. Submitted by a physician who both initiates and conducts an investigation, and under whose immediate direction the investigational drug is administered or dispensed. Goal is to study: an unapproved drug an approved product for a new indication or in a new patient population dietary supplements – may be considered drugs Types of IND applications
New Full Committee Clinical Application Form Financial Sponsor: Private Co, Foundation, Feds, individual donations, PI’s Ed fund, Department Regulatory Sponsor Who prepared the protocol Is the study exempt? If the study is initiated by a UC Davis Principal Investigator and the study is NOT exempt: I understand that I am a regulatory sponsor and investigator on this study, assuming responsibility for all regulatory requirements specified in 21 CFR 312 (Investigational New Drug Application) and/or 21 CFR 812 (Investigational Device Exemption). FDA Form 1572 is attached (required for regulatory sponsor-investigators) What is “Regulatory Sponsor”?
Expanded Access Emergency Use IND “Compassionate Use” experimental drug in an emergency situation that does not allow time for submission of a “regular” IND. Also used for patients who do not meet the criteria of an existing study protocol, or if an approved study protocol does not exist Treatment IND: promising experimental drugs for serious or immediately life-threatening conditions while FDA review is ongoing Types of IND applications
FDA = no formal compassionate use policy Patient does not meet existing protocol eligibility Life-threatening situation Company agrees to ship the drug Exempt from prior IRB review and approval IRB notified within 5 days Verbal approval by the FDA by phone Written follow-up Informed Consent required Company required to follow up Emergency IND (Compassionate Use)21 CFR 312.36
Facilitate availability of drugs to critically ill patients, even if the drug is not yet approved Available to patients not enrolled in controlled trials No comparable or satisfactory treatments The drug is currently in a controlled clinical trial under an IND in Phase III (for immediately life-threatening diseases - in phase II) PI can submit separate treatment IND, providing that the drug manufacturer agreed to provide the drug and to authorize incorporation-by-reference of the technical information required for treatment IND. Treatment IND21 CFR 312.34 and .35
Agreement between FDA and National Cancer Institute (NCI) Group C classification system allows access to certain cancer drugs Group C drugs: generally Phase III - have shown reproducible anti-tumor activity Provided only to properly trained physicians who have registered themselves with NCI Group C drugs are provided free of charge See Handout Group C Treatment IND
Letter of Authorization provided by Sponsor/Drug Manufacturer Authorize incorporation-by-reference of the technical information required for treatment IND See Example Treatment IND (cont.)
Applies to all clinical investigations of products that are subject to section 505 of the Federal Food, Drug, and Cosmetic Act or to the licensing provisions of the Public Health Service Act (58 Stat. 632, as amended (42 U.S.C. 201 et seq .)). New molecular entity Lawfully marketed drug/biologics for a new indication, new formulation or in a new combination (with exceptions) Investigation compound (under IND from a company) See decision tree When is an IND Required?
Not intended to be reported to FDA as a well-controlled study in support of a new indication Not intended to support a significant change in the advertising/labeling for the product Does not involve a route of administration or dosage level or use in a patient population or other factor that significantly increases the risks (or decreases the acceptability of the risks) of the drug product IND Exemptions for lawfully marketed drugs21 CFR 312.2(b)(1)
Conducted in compliance with the requirements for institutional review As per part 56 and requirements for informed consent in part 50 Conducted in compliance with requirements of Sec. 312.7 (i.e., not promoting or charging for investigational drugs ) IND Exemptions for lawfully marketed drugs 21 CFR 312.2(b)(1) (continued)
Investigator reviews requirements for exemption per 21 CFR 312.2(b)(1) Guidance for Industry IND Exemptions for Studies of Lawfully Marketed Drugs or Biological Products for the Treatment of Cancer Clinical Trials Navigator (or designee) confirms that each requirement for exemption is addressed Exemption items reviewed with study team at Site Initiation Visit Consensus obtained Memorandum on UCD letterhead [Investigator and Co-Investigator sign off] See Examples IND Exemption Review Process: The UCD Cancer Center
Overview of IND Process for academics FDA will send meeting minutes and recommendations FDA will respond with the date 30 days 14 days 60 days Prepare and submit IND Request pre-IND mtg Pre-IND mtg Pre-IND materials due 4 weeks prior
Overview of IND Process for academics IND effective date 1 year date 60 days 30 days Prepare and submit IND Annual Report Due Protocol Amendments Information Amendments IND Safety Reports
21 CFR 312.38 Sponsor may withdraw IND at any time w/o prejudice Notify FDA of the reasons End clinical investigations All drug stock disposed of or returned to the drug manufacturer Withdrawal of IND/ Inactive Status • 21 CFR 312.45 • On clinical hold for > 1 year • No subjects enrolled for >2 years • FDA will notify the sponsor – needs response within 30 days • To reopen – sponsor must submit a protocol amendment – wait for 30 days • Terminated after 5 years in inactive status
FDA proposes termination giving opportunity to respond Clinical investigations are being conducted in a manner substantially different than that described in the protocols submitted in the IND. Drug is being promoted or distributed for commercial purposes not justified by the requirements of the investigation or permitted by §312.7. Termination by the FDA21 CFR 312.44
IND, or any amendment or report to the IND, contains an untrue statement of a material fact or omits material information required by this part. !Sponsor fails promptly to investigate and inform the FDA and all investigators of serious and unexpected adverse experiences !Sponsor fails to submit an accurate annual report Termination by the FDA (continued)21 CFR 312.44
21 CFR 312 Subpart D Collect Investigator CVs Ensure Investigators have current protocol version Aware of any adverse events associated with the drug Control of drug under investigation FDA Form 1572 – ensure investigation is conducted according to signed Statement of the Investigator IRB approvals – Informed Consent for each subject Protect RIGHTS, SAFETY, and WELFARE of subject General Responsibilities under IND
12 Clinical Research Coordinators 3 Oncology Clinical Nurses 5 Regulatory Coordinators 2 Database Administrators Clinical Trials Navigator Clinical Trials search website http://ccresources.ucdmc.ucdavis.edu/csr/ content/clinicaltrialspublicsearch.csr Cancer Center: Clinical Trials Support Unit (CTSU)
Cancer Therapy Evaluation Program (CTEP) – National Program for Cancer Research Suggested Templates [Phase I, II, III] http://ctep.cancer.gov/protocolDevelopment/templates_applications.htm#policiesAndGuidelines See Handout – Detailed Table of Contents Protocol Development
SCHEMA 1. OBJECTIVES 2. BACKGROUND 3. PATIENT SELECTION 4. REGISTRATION PROCEDURES 5. TREATMENT PLAN 6. DOSING DELAYS/DOSE MODIFICATIONS 7. ADVERSE EVENTS: LIST AND REPORTING REQUIREMENTS 8. PHARMACEUTICAL INFORMATION 9. CORRELATIVE/SPECIAL STUDIES 10.STUDY CALENDAR 11.MEASUREMENT OF EFFECT 12.DATA REPORTING / REGULATORY CONSIDERATIONS 13.STATISTICAL CONSIDERATIONS REFERENCES INFORMED CONSENT TEMPLATE APPENDICES Protocol Development –Table of Contents
Subjects may not be enrolled onto a new study until a protocol initiation meeting has taken place Completion of the Study Initiation Checklist and the Site Signature and Responsibility Log The completed checklist and log filed in the study regulatory files As study personnel change, the Regulatory Coordinator will update the Site Signature and Responsibility Log to reflect these personnel changes Protocol Initiation Meeting aka “Site Initiation Visit”
Members of the research team attend a study-specific initiation meeting within four weeks of study activation or before enrolling the first subject. [Clinical Research Coordinator (CRC), Regulatory Coordinator, study nurse, Principal Investigator (PI)], attendees from the Investigational Drug Service and the Cancer Center Pharmacy, and other interested parties If the PI is unavailable, he/she may appoint another study investigator to attend on his/her behalf. Protocol Initiation Meetingaka “Site Initiation Visit”
At a minimum, study-specific initiation meetings will include a review of: research team member responsibilities eligibility criteria treatment schedule pretreatment procedures AE reporting requirements pending revisions/modification/amendments study agents to be ordered study-specific supplies to be ordered The completed Study Initiation Checklist and the signed Site Signature and Responsibility Log will serve as documentation that the initiation meeting occurred. Protocol Initiation Meeting aka “Site Initiation Visit”
For investigator-initiated studies, the PI and the CRC meet monthly, at a minimum, to discuss the status of the study. If study involves dose escalation, the dose limiting toxicity (DLT) status of the current cohort of subjects is discussed. Based on these subjects, the PI will decide whether or not to escalate the dose level. The CRC assigned to the study creates minutes for all study-related meetings with the PI. If study involves dose escalation, the CRC assigned to the study will complete the Dose Escalation Minutes form. This is filed in the study’s regulatory file, and one copy will be submitted to the Phase I Committee coordinator. Investigator-Initiated Study-Specific Meetings
Must register study within 21 days of 1st patient enrollment Using Protocol Registration System (PRS) http://prsinfo.ClinicalTrials.gov – set up organization account Denise Owensby – PRS Administrator – for account set up Can modify study information See Handout Post Study on http://www.ClinicalTrials.gov
Thank you for your attention. Questions? Clarification? The end