Download

1 / 27
310 likes | 468 Views
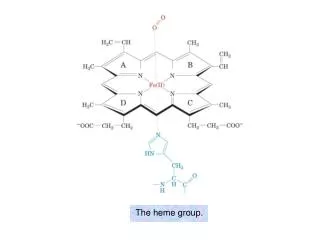
Disorders of Heme synthesis. HEME-CONTAINING PROTEINS Hemoglobin Myoglobin Cytochromes Catalase Some peroxidases. STRUCTURE OF HEME. Ferrous iron (Fe 2+ ) Protoporphyrin IX: contains 4 pyrrole rings linked together by methenyl bridges.

E N D
HEME-CONTAINING PROTEINS Hemoglobin Myoglobin Cytochromes Catalase Some peroxidases
STRUCTURE OF HEME Ferrous iron (Fe2+) Protoporphyrin IX: contains 4 pyrrole rings linked together by methenyl bridges • The two major cell types that are active in heme synthesis are hepatocytes and bone marrow erythroblasts • 85% of total synthesis occurs in erythroid cells • 80% of liver production is used for cytochromes
Disorders of Heme metabolism Heme biosynthesis Porphyrias Heme degradation Jaundice
Stercobilin excreted in feces Urobilin excreted in urine Hemoglobin Globin Heme Urobilinogen formed by bacteria O2 Heme oxygenase CO Biliverdin IX NADPH Bilirubin diglucuronide (water-soluble) Biliverdin reductase NADP+ 2 UDP-glucuronic acid Bilirubin (water-insoluble) Bilirubin (water-insoluble) via blood to the liver BLOOD CELLS KIDNEY reabsorbed into blood INTESTINE via bile duct to intestines LIVER Figure 2. Catabolism of hemoglobin
The Porphyrias • Group of inherited or acquired disorders of heme production • 8 enzymes in heme biosynthetic pathway • First and the last 2 are mitochondrial, while the other five are in the cytosol.
Classification of the Porphyrias • Multiple ways to categorize porphyrias: • Hepatic vs. Erythropoietic: Organ in which accumulation of porphyrins and their precursors appears • Cutaneous vs. Non- cutaneous • Acute and non-acute forms • Acute: • Aminolevulinate dehydratase deficiency porphyria (ALA-D) • Acute intermittent porphyria (AIP) • Hereditary coproporphyria (HCP) • Variegate porphyria (VP) • Chronic: • Porphyria cutanea tarda (PCT) • Erythropoietic protoporphyria (EPP) • Congenital erythropoietic porphyria (CEP) • Hepatoerythropoietic porphyria (HEP)
Enzymatic Deficiencies • All of the heme pathway intermediates are potentially toxic. • Their overproduction causes the characteristic neurovisceral and/or photosensitizing symptoms.
PORPHYRIA CUTANEA TARDA • Most common porphyria • Hepatic, autosomal dominant • Disease is caused by a deficiency in uroporphyrinogen decarboxylase, which is involved in the conversion of uroporphyrinogen III to coproporphyrinogen III • Uroporphyrinogen accumulates in urine • Patients are photosensitive (cutaneous photosensitivity) • Accumulation of porphyrinogens results in their • conversion to porphyrins by light • Porphyrins react with molecular oxygen to form • oxygen radicals • Oxygen radicals can cause severe damage to the • skin
Acute intermittent porphyria Van Gogh King George III Mary Queen of Scots
Acute intermittent porphyria • The prevalence of AIP in the United States is thought to be 5–10 per 100 000. • It is more common in northern European countries, such as Sweden (60–100 per 100 000), Britain and Ireland. • Acute intermittent porphyria PBGD gene mutation is inherited in an autosomal dominant fashion. • Affects women more than men, with a ratio of 2:1. • Most patients become symptomatic at age 18-40 years. • Attacks occurring before puberty or after age 40 years are unusual unless a major provocation • Most patients are completely free of symptoms between attacks. • Course of the neurological manifestations is highly variable. • Acute attacks of porphyria may resolve quite rapidly. • Sudden death may occur, presumably due to cardiac arrhythmia.
Symptoms • Attacks involve neuro-visceral symptoms but no skin manifestations: • The sequence of events in attacks usually is (1) abdominal pain, (2) psychiatric symptoms, such as hysteria, and (3) peripheral neuropathies, mainly motor neuropathies. • Gastroenterological Symptoms most common: • Constipation (48–84%), colicky abdominal pain (occurring in 85–95% cases), vomiting (43–88%), diarrhea (5–12%) • Patients may have CNS signs consisting of seizures (10–20%), mental status changes, cortical blindness, and coma. • Patients often experience peripheral neuropathies (42–60%) that are predominantly motor and can mimic Guillain-Barré syndrome. • Patients may develop fever(9–37%), hypertension (36–54%) and tachycardia (28–80%).
Mechanism • The exact mechanism underlying these complaints is not yet well understood, various hypotheses have been put forward: • Excess amounts of PBG or ALA may cause neurotoxicity (Meyer et al, 1998) • Increased ALA concentrations in the brain may inhibit gamma-aminobutyric acid release (Mueller & Snyder, 1977; Brennan & Cantrill, 1979) • Heme deficiency may result in degenerative changes in the central nervous system (Whetsell et al, 1984) • Decreased heme synthesis in the liver results in decreased activity of hepatic tryptophan pyrrolase (TP), a heme-dependent enzyme, possibly resulting in increased levels of serotonin
Precipitants • Drugs: most common precipitate of acute attacks : • Barbiturates and sulphonamides being most common • Reduced energy intake: even brief periods of starvation during dieting, postoperative periods, or concurrent illness. • Tobacco smoke: polycyclic aromatic hydrocarbons, are known inducers of hepatic cytochrome P450 enzymes and heme synthesis. • An association between cigarette smoking and repeated attacks of porphyria was found in a survey of 144 patients with AIP in Britain (Lip et al, 1991). • Infections, surgery and stress.
Diagnosis • Demonstration of porphyrin precursors, such as ALA and/or PBG, is essential for the diagnosis of acute porphyrias. • Porphyrin analysis is necessary for the diagnosis of porphyrias with cutaneous photosensitivity. • PBG usually is not included in a urine porphyrin screen and must be ordered specially • Molecular diagnostic testing: • Detection of PBGD mutations in AIP provides 95% sensitivity and around 100% specificity • Possible to screen asymptomatic gene carriers. • Less Useful in acute attacks PBG in urine is oxidized to porphobilin upon standing, which gives a dark-brown color to urine, and often referred to as ‘port-wine reddish urine’.
It is the most common childhood porphyria. • It is usually evident by 2 years of age. Pathogenesis deficient activity of ferrochelatase enzyme Erythropoietic Protoporphyria
Lab. finding: • Plasma porphyrin level and fluorescence spectrum • Increased free protoporphyrin in RBCs, stool • CBC, LFTs • Liver/gallbladder imaging
Congenital Erythropoietic porphyria ( Gunther's disease ): It is a very rare autosomal recessive disorder. Patients usually present during infancy and rarely present in adult life with milder forms. Pathogenesis It is caused by elevation of both water-soluble and lipid-soluble porphyrin levels due to deficiency of uroporphyrinogen III synthase enzyme. Clinical features Very severe photosensitivity with phototoxic burning and blistering leading to mutilation of light exposed parts. Erythrodontia. Scleromalacia perforans. Hypersplenism. Hemolytic anemia. Thrombocytopenia Lab. finding Uroporphyrin and Coproporphyrin in urine Corproporphyrin in stool
Hepatoerythropoietic Porphyria • Inheritance/Pathogenesis: • AD • Uroporphyrinogen (UROGEN) decarboxylase deficient • Incidence: • Very rare -Presents at age 1 • Prognosis: • Normal life span Clinical picture: • Skin • Similar to CEP—Severe photosensitivity with burning, edema, vesicles/bullae, erosions, infection • Late changes—Mutilating scars with deformation of nose, ears, fingers; scarring alopecia, pigmentary changes, sclerodermoid changes • Hypertrichosis • Teeth • Red/brown color • Eyes • Photophobia, ectropion, conjunctivitis
Heme • Hemolytic anemia • GI • Splenomegaly • GU • Dark urine at birth • Plasma porphyrin level and fluorescence spectrum • protoporphyrin in RBCs • urinary uroporphyrin • fecal coproporphyrin • CBC Lab. finding:
Varigeate Porphyria • Inheritance: • AD • Protoporphyrinogen oxidase gene (PROTOGEN) • Severe forms associated with hemochromatosis gene • Prenatal Diagnosis: • DNA analysis • Incidence: • Most common in South African whites 1:330 • Elsewhere is 1:50,000 to 100,000 • M=F • Age at Presentation: • Begins after puberty in second and third decade of life • Pathogenesis: • Mutation in PROTOGEN oxidase gene causes a 50% decrease in PROTOGEN oxidase activity • Acute attacks precipitated by: • Drugs: barbiturates, estrogen, griseofulvin, sulfonamides • Infection • Fever • Alcohol • Pregnancy • Decreased caloric intake • Increase Δ-aminolevulinic acid (ALA) synthetase with attacks
Clinicalpicture: • Skin: • Identical to PCT with bullae, erosions, skin fragility, scarring, hypertrichosis, hyperpigmentation on photodistributed face, neck and dorsum of hands • Acute Attacks (i.e., Acute Intermittent Porphyria and Hereditary Coproporphyria): • Gastrointestinal: • Colickly abdominal pain, nausea, vomiting, constipation • CNS: • Peripheral neuropathy with pain, weakness, paralysis • Confusional state, anxiety, depression, delerium • Seizures, coma • CV: • Tachycardia, hypertension
Laboratory Data: • Plasma porphyrin level • Plasma porphyrin fluorescence spectrum—626 nm is diagnostic • 24 hour urine porphyrin levels: coproprophyrin = or > uroporphyrin • Urine ALA and porphobillinogen (PBG) levels increased during attacks • Fecal prophyrin levels: markedly elevated, protoporphyrin>coproporphyrin