Download

1 / 26
260 likes | 286 Views
Learn about homology modeling, a method using known protein sequences to predict 3D structures. Understand the process, why it's crucial, and its advantages. Step-by-step guide with tools and tips for accurate predictions.

E N D
Tutorial Homology Modelling
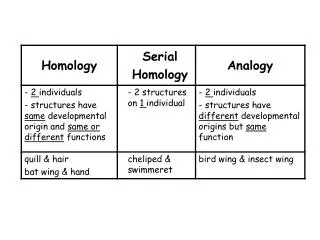
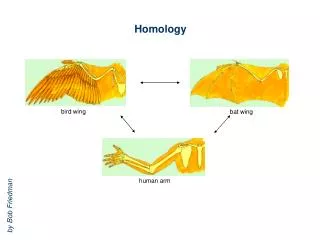
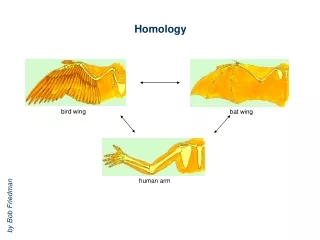
Sequence-Structure-Function Relationships • Proteins of similar sequences fold into similar structures and perform similar biological functions. • The protein sequence has the intrinsic information to encode the protein structure.
The Noble Prize in Chemistry 1972 Christian B Anfinsen "for his work on ribonuclease, especially concerning the connection between the amino acid sequence and the biologically active conformation"
The protein sequence is sufficient to specify its 3D structure From Nobel Lecture, December 11, 1972, by Christian Anfinsen
Sequence->Structure->Function • Widespread Automated DNA sequencing => more sequence data than structure data • Semi-Automated pipeline of structure determination is still not widespread. • Nevertheless, structure is more conserved than sequence. • Sequence homologs => structural homologs • See Chapter 9, Baxevanis and Ouellette 3rd edn.
Protein Structure Prediction vs Experimental Determination From Chapter 9, Bryan Bergeron, Bioinformatics Computing, 2003 Pearson Education, Inc.
Structure Prediction from sequence • Homology (or comparative) modelling • Threading • Ab initio calculationsHomology modelling is most accurate and powerful
What is Homology Modeling? • Homology modeling also known as comparative modeling uses homologous sequences with known 3D structures for the modelling and prediction of the structure of a target sequence. • Homology modeling is one of the most best performing prediction methods that gives “accurate” predicted models.
How is Homology Modeling done • Multistep process involves many steps such as: • Sequence alignment of target/query/unknown protein sequence to homologous sequence with a known structure • structure modification of backbone • side chain replacements • Energy minimisation for refinement of structural model • Validation of model with visual inspection and etc
Why Homology Modeling? • The number of protein structures solved so far are fewer than the number of genes known. • Proteins of biological interest with their orthologous proteins solved by X-ray crystallography or NMR can be modeled. • Homology modeling is an important method used to predict the structures of membrane proteins, ion channels, transporters that are large and difficult to crystallize. • Examples: GPCR (G Protein-coupled receptor), cytochrome P450 etc.
Overview of the process of Homology Modeling • A target sequence (the structure to be predicted) • Identify the homologous sequence with known 3D as template • Using homology modeling software such as Modeller for structure prediction (from the Sali Lab) • Model evaluation and refinement
Pre-Modeling Stage: Template Identification • Target sequence in FASTA format as input • Blastp against PDB • Identify proteins with “good” hit • Pairwise or multiple sequence alignment • Further editing the alignment results • Realign and identify the “good” structural template
Pre-Modeling Stage: Preparing the Input Files for Modeller • PDB files for structural templates is required • The PIR file from the alignment results • The script file model.top to execute the Modeller program(latest versions use Python scripts)
In the Heart of Modeller From the Modeller manual
Evaluation of Predicted ModelGarbage in-Garbage out • The predicted model can be superimposed with known structure determined by experiment http://wishart.biology.ualberta.ca/SuperPose/ • The predicted model is normally evaluated by root mean square deviation (RMSD)
Calculating RMSD • N = number of atoms, d = the distance in Angstrom between corresponding atoms in the experimental and predicted protein structures. From Chapter 9, Bryan Bergeron, Bioinformatics Computing, 2003 Pearson Education, Inc.
Some Rule of Thumb for Structural Modelling • Proteins that share 35 to 50% sequence identity with their templates, will generally deviate by 1.0 to 1.5 Å from their experimental counter parts. • Crystallographic structures of identical proteins can vary not only because of experimental errors and differences in data collection conditions and refinement, but also because of different crystal lattice contacts and the presence or absence of ligands.
Quality of Model • The correctness of a model is essentially determined by the quality of the sequence alignment used to identify the template. • If the sequence alignment is wrong in some regions, then the spatial arrangement of the residues in this portion of the model will be incorrect.
Viewing the Model • The predicted model is saved in PDB format that can be viewed by molecular visualizing software such as Rasmol, PyMol, MolMol, Sybyl etc. • Viewing is an essential step to validate the quality of the predicted model. • In this practical, Rasmol is used to view the predicted structure.
Model Refinement • Gaps in sequence alignment represent insertion/deletion regions of target. Loop modeling is used to refine these regions (not cover in this practical) • The predicted model can be further refined by energy minimization to remove unfavourable non-bonded contacts with force fields such as CHARMM, AMBER or GROMOS etc (not covered in this practical)
Web-Based Homology Modeling: The SWISS-MODEL Server • The aim of the Internet-based SWISS-MODEL server is to provide a comparative protein modelling tool independent from expensive computer hardware and software. http://www.expasy.ch/swissmod/SWISS-MODEL.html
Steps involved in SwissModel http://swissmodel.expasy.org/ • Take target sequence of unknown structure • Using BLAST to select closest homolog with known structure as structural template http://swissmodel.expasy.org/SM_Blast.html • Insert target sequence and homologous sequence to Web service http://swissmodel.expasy.org/SM_FIRST.html • Results will be emailed back to you. • Warning: Structure needs to be analysed and validated
Simple Homology Modelling using Modeller • Take target sequence of unknown structure • Using BLAST to select closest homolog with known structure. • Using Clustalx or Jalview to do pairwise alignment between target sequence and structural homolog and manual adjustment • Inspection of missing structural features in structural homolog • Preparation of alignment file align.pir • Use Modeller7v7 software (http://salilab.org/modeller/) to do the homology modelling
Structure Validation • Visual inspection • Minimise torsion angles in disallowed regions of Ramachandran plots • Maximised hydrogen bonding • Minimised exposed hydrophobic residues • Packing etc. • Analysis – e.g. run Procheck (http://www.biochem.ucl.ac.uk/~roman/procheck/procheck.html), VADAR, Verify3D etc