Download

1 / 43
550 likes | 919 Views
Homology modeling. Dinesh Gupta ICGEB, New Delhi. Protein structure prediction. Methods: Homology (comparative) modelling Threading Ab-initio. Protein Homology modeling.

E N D
Homology modeling Dinesh Gupta ICGEB, New Delhi
Protein structure prediction • Methods: • Homology (comparative) modelling • Threading • Ab-initio
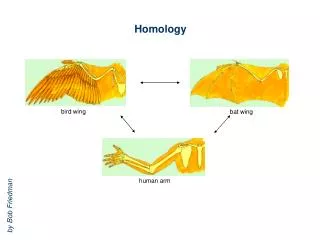
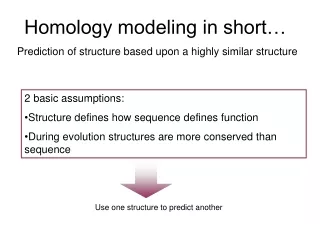
Protein Homology modeling • Homology modeling is an extrapolation of protein structure for a target sequence using the known 3D structure of similar sequence as a template. • Basis: proteins with similar sequences are likely to assume same folding • Certain proteins with as low as 25% similarity have been observed to assume same 3D structure
The accuracy of modeling is proportional to the similarity in primary sequences
Steps… • Given: • A query sequence Q • A database of known protein structures • Find protein P such that P has high sequence similarity to Q • Return P’s structure as an approximation to Q’s structure • Energy minimization
Sofware for homology molecular modelling • Freeware: available for all OS • Downloadable • Modeller (Sali, 1998) • DeepView (SwissPDB viewer) • WHATIF (Krieger et al. 2003) • Web based: • SWISS MODEL server (www.expasy.org/swissmod/SWISS-MODEL.html) • CPH model server (http://www.cbs.dtu.dk/services/CPHmodels) • SDSC1 server (http://cl.sdsc.edu/hm.html)
Protein structure prediction • Methods: • Homology (comparative) modelling • Threading • Ab-initio
Threading • Structure prediction that picks up where homology modelling leaves off. • Recognize folds in proteins having no similarity to known proteins structures • Very approximate models • Check by forcing a sequence of structure into known folds checking the packing of aa residues, including sides chains, in each fold.
2 kinds of threading • Three dimensional threading • Distance Based Method (DBM) • Two dimensional threading • Prediction Based Methods (PBM)
Threading software • EVA: http://cubic.bioc.columbia.edu/eva/ • SAMt99: http://www.cse.ucsc.edu/research/compbio/HMM-apps/T99-model-library-search.html • 3DPSSM: http://www.sbg.bio.ic.ac.uk/3dpssm • FUGUE: http://tardis.nibio.go.jp/fugue/ • Metaservers:
Protein structure prediction • Methods: • Homology (comparative) modelling • Threading • Ab-initio
Ab initio structure prediction • Still experimental • ROSETTA (David Baker)
Energy minimization (Molecular Mechanics, MM) • Energy minimization is an important part of both empirical and predicted structures • MM could be used to calculate large scale conformational changes over long periods of time, but currently computationally infeasible.
How does MM work? • Three aspects: • Functions that describe the forces acting on the atoms • Numerical integration methods, to calculate the motion of the atoms due to the forces acting on them • Long time propagation of the equations of motion • Computational demands are intense • Accuracy (small errors propagate!) • Stability • Lots of techniques for approximation (e.g. rigid bodies) and handling artifacts (resonance).
The Force Fields • How do atoms stretch, vibrate, rotate, etc.? • Must represent the constraints on atomic motion (e.g. van der Waals, electrostatic, bonds, etc.) • Must also represent solvation effects etc. • Quantum solutions exist, but are too complex to calculate for such large systems • Empirical (approximate) energy functions must be used. No single best function exists.
Real energetics • Steric (conformational) energy. Additive combination of • Bonded: stretching, bending, stretching and bending • Non-bonded: Van der Waals, electrostatic and “torsional” • Minimum energy conformation minimizes these energies • Rosetta energy function is an empirical attempt to capture most of this energy function without having to calculate it fully.
Bond length • Spring-like term for energy based on distance Estr = ½ks,ij(rij -ro)2where ks,ij is the stretching force constant for the bond between i and j, rij is the length, and ro is the equilibrium bond length
Bond bend • Same basic idea for bendingEbend = ½kb,ij(ij –o)2where where kb,ij is the bending force constant, ij is the instantaneousbond angle, and o is the equilibrium bond angle
Stretch-bend • When a bond is bent, the two associated bond lengths increase, with interaction term: Estr-bend =½ksb,ijk(rij-ro)(ik - o)where ksb,ijk is the stretch-bend force constant for the bond betweenatoms i and j with thebend between atomsi, j, and k.
Van der Waals • A non-bonded interaction capturing the preferred distance between atomswhere A and B are constants depending on the atoms. For two hydrogen atoms, A=70.4kCÅ6 and B=6286kCÅ12
Electrostatics • If bonds in the molecule are polar, some atoms will have partial electrostatic charges, which attract if opposite and repel otherwise. where Qi and Qj are the partial atomic charges for i and j separated by distance rij , is the dielectric constant of the solute, and k is a units constant (k=2086 kcal/mol)
Torsional energy • Torsion is the energy needed to rotate about bonds. Only relevant to single bonds, since others are too stiff to rotate at all Etor = ½ktor,1 (1 - cos ) + ½ktor,2 (1 - 2cos ) + ½ktor,3 (1 - 3cos )where is the dihedral anglearound the bond, and ktor,1, ktor,2and ktor,3 are constants for one-,two- and three-fold barriers. energy of 3-fold torsionalbarrier in ethane
Energy minimization • Given some energy function and initial conditions, we want to find the minimum energy conformation. • Optimization problem, various methods: • Steepest descent • Conjugate gradient descent • Newton-Raphson • Various programs: Charmm, Amber are two most widely used (and packaged)
Time steps Need time steps of roughly 1/10 the period of the smallest time scale of interest, or about a femtosecond (10-15s). A million computational steps per nanosecond of simulation...
Issues in Molecular Mechanics • Solvation models: water & salt are very important to molecular behaviour. Must model as many water atoms as protein atoms. • Initial conditions: velocity & position • Equilibration: simulated heating and cooling • Chaos: sensitivity to initial conditions, and statistical characterization of states • Computational issues (e.g. parallelization)
Molecular Dynamics • Molecules, especially proteins, are not static. • Dynamics can be important to function • Trajectories, not just minimum energy state. • MM ignores kinetic energy, does only potential energy • MD takes same force model, but calculates F=ma and calculates velocities of all atoms (as well as positions)
Docking • Computation to assess binding affinity • Looks for conformational and electrostatic "fit" between proteins and other molecules e.g. inhibitors • Optimization again: what position and orientation of the two molecules minimizes energy? • Large computations, since there are many possible positions to check, and the energy for each position may involve many atoms
Virtual Screening • Docking small ligands to proteins is a way to find potential drugs. Industrially important • A small region of interest (pharmacophore) can be identified, reducing computation • Empirical scoring functions are not universal • Various search methods: • Rigid provides score for whole ligand (accurate) • Flexible breaks ligands into pieces and docks them individually
Docking example Benzamidine binding to beta-Trypsin 3ptb,
Macromolecular docking • Docking of proteins to proteins or to DNA • Important to understanding macromolecular recognition, genetic regulation, etc. • Conceptually similar to small molecule docking, but practically much more difficult • Score function can't realistically compute energies • Use either shape complementarities alone or some kind of mean field approximation
Docking Resources • AutoDock http://www.scripps.edu/pub/olson-web/doc/autodock/ • FlexX http://www.biosolveit.de/FlexX/ and commercially at http://www.tripos.com • Dock http://www.cmpharm.ucsf.edu/kuntz/dock.html • 3D-Dock http://www.bmm.icnet.uk/docking/ which uses an unusual “Fourier correlation” method and is aimed at protein-protein interactions
Lab Exercise-1 Install: • MDL chime • RasMol • SwissPDBviewer • Cn3D Explore few protein/DNA structures
Lab exercise-2 • Download sequence file for S. cerevisiae endoplasmic reticulum mannosidase • Generate a homology model using SWISS-model server http://www.expasy.ch/swissmod/ • Download the template structure from www.rcsb.org • Compare the model and template structures • Repeat the exercise for other protein sequences of your choice