Download
1 / 1
20 likes | 204 Views
Inference of Allele S pecific Expression Levels from RNA- Seq Data. H0. H1. Sahar Al Seesi and Ion M ă ndoiu. Allele Specific Gene/Isoform Expression. Make cDNA & shatter into fragments. Computer Science and Engineering Dept., University of Connecticut.

E N D
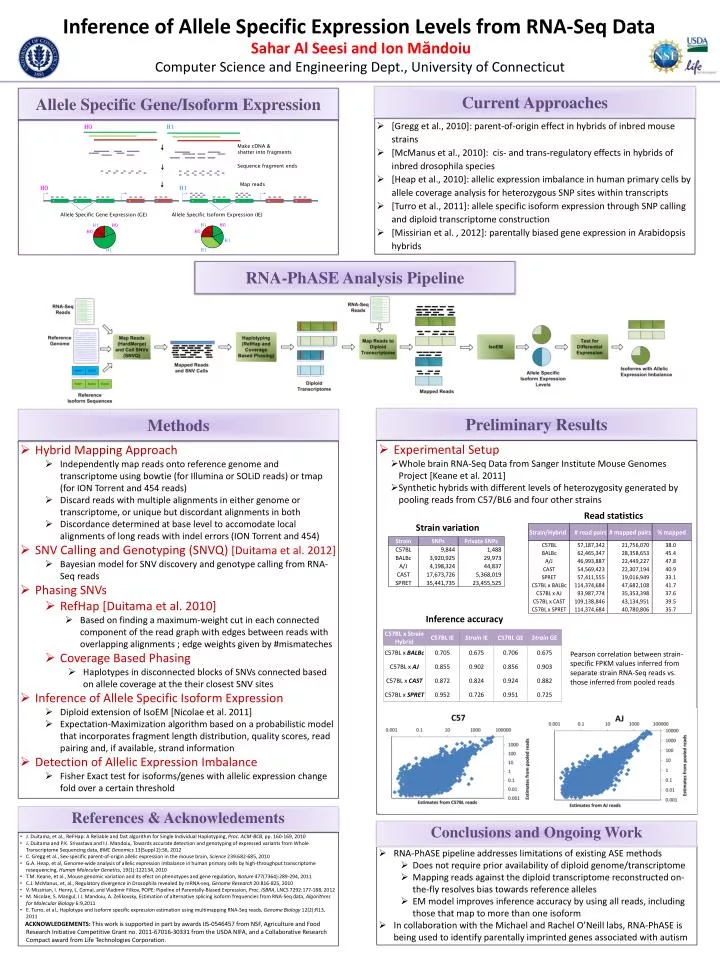
Inference of Allele Specific Expression Levels from RNA-Seq Data H0 H1 Sahar Al Seesi and Ion Măndoiu Allele Specific Gene/Isoform Expression Make cDNA & shatter into fragments Computer Science and Engineering Dept., University of Connecticut Sequence fragment ends Map reads H0 H1 Current Approaches A A B B C C D D E E Allele Specific Gene Expression (GE) Allele Specific Isoform Expression (IE) • [Gregg et al., 2010]: parent-of-origin effect in hybrids of inbred mouse strains • [McManus et al., 2010]: cis- and trans-regulatory effects in hybrids of inbred drosophila species • [Heap et al., 2010]: allelic expression imbalance in human primary cells by allele coverage analysis for heterozygous SNP sites within transcripts • [Turro et al., 2011]: allele specific isoform expression through SNP calling and diploid transcriptome construction • [Missirian et al. , 2012]: parentally biased gene expression in Arabidopsis hybrids RNA-PhASE Analysis Pipeline Preliminary Results Methods • Experimental Setup • Whole brain RNA-Seq Data from Sanger Institute Mouse Genomes Project [Keane et al. 2011] • Synthetic hybrids with different levels of heterozygosity generated by pooling reads from C57/BL6 and four other strains • Read statistics • Strain variation • Inference accuracy • Hybrid Mapping Approach • Independently map reads onto reference genome and transcriptome using bowtie (for Illumina or SOLiD reads) or tmap (for ION Torrent and 454 reads) • Discard reads with multiple alignments in either genome or transcriptome, or unique but discordant alignments in both • Discordance determined at base level to accomodate local alignments of long reads with indel errors (ION Torrent and 454) • SNV Calling and Genotyping (SNVQ) [Duitama et al. 2012] • Bayesian model for SNV discovery and genotype calling from RNA-Seq reads • Phasing SNVs • RefHap [Duitama et al. 2010] • Based on finding a maximum-weight cut in each connected component of the read graph with edges between reads with overlapping alignments ; edge weights given by #mismateches • Coverage Based Phasing • Haplotypes in disconnected blocks of SNVs connected based on allele coverage at the their closest SNV sites • Inference of Allele Specific Isoform Expression • Diploid extension of IsoEM [Nicolae et al. 2011] • Expectation-Maximization algorithm based on a probabilistic model that incorporates fragment length distribution, quality scores, read pairing and, if available, strand information • Detection of Allelic Expression Imbalance • Fisher Exact test for isoforms/genes with allelic expression change fold over a certain threshold Pearson correlation between strain-specific FPKM values inferred from separate strain RNA-Seq reads vs. those inferred from pooled reads References & Acknowledements Conclusions and Ongoing Work • J. Duitama, et al., ReFHap: A Reliable and fast algorithm for Single Individual Haplotyping, Proc. ACM-BCB, pp. 160-169, 2010 • J. Duitama and P.K. Srivastava and I.I. Mandoiu, Towards accurate detection and genotyping of expressed variants from Whole Transcriptome Sequencing data, BMC Genomics 13(Suppl 2):S6, 2012 • C. Gregg et al., Sex-specific parent-of-origin allelic expression in the mouse brain, Science 239:682-685, 2010 • G.A. Heap, et al, Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptomeresequencing,Human Molecular Genetics, 19(1):122134, 2010 • T.M. Keane, et al., Mouse genomic variation and its efect on phenotypes and gene regulation, Nature 477(7364):289-294, 2011 • C.J. McManus, et, al., Regulatory divergence in Drosophila revealed by mRNA-seq, Genome Research20:816-825, 2010 • V. Missirian, I. Henry, L. Comai, and Vladimir Filkov, POPE: Pipeline of Parentally-Biased Expression, Proc. ISBRA, LNCS 7292:177-188, 2012 • M. Nicolae, S. Mangul, I.I. Mandoiu, A. Zelikovsky, Estimation of alternative splicing isoform frequencies from RNA-Seq data, Algorithms for Molecular Biology 6:9,2011 • E. Turro, et al., Haplotype and isoform specific expression estimation using multimapping RNA-Seq reads, Genome Biology12(2):R13, 2011 • ACKNOWLEDGEMENTS: This work is supported in part by awards IIS-0546457 from NSF, Agriculture and Food Research Initiative Competitive Grant no. 2011-67016-30331 from the USDA NIFA, and a Collaborative Research Compact award from Life Technologies Corporation. • RNA-PhASEpipeline addresses limitations of existing ASE methods • Does not require prior availability of diploid genome/transcriptome • Mapping reads against the diploid transcriptomereconstructed on-the-fly resolves bias towards reference alleles • EM model improves inference accuracy by using all reads, including those that map to more than one isoform • In collaboration with the Michael and Rachel O’Neill labs, RNA-PhASE is being used to identify parentally imprinted genes associated with autism