Download

1 / 27
270 likes | 405 Views
Association Tests for Rare Variants Using Sequence Data. Guimin Gao , Wenan Chen, & Xi Gao Department of Biostatistics, VCU. Introduction to Association tests: two hypotheses. Common variant-common disease Common variant: Minor allele frequencies (MAF) >= 5%

E N D
Association Tests for Rare Variants Using Sequence Data GuiminGao, Wenan Chen, & Xi Gao Department of Biostatistics, VCU
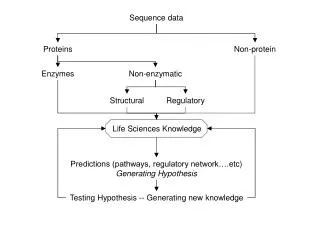
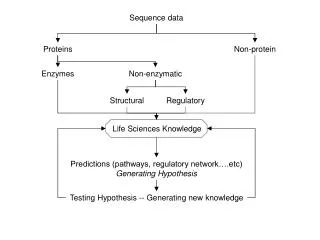
Introduction to Association tests: two hypotheses • Common variant-common disease • Common variant: Minor allele frequencies (MAF) >= 5% • Using linkage disequilibrium(LD) • Rare variant-common disease • Rare variant: MAF < 1% (or 5%) • High allelic heterogeneity: collectively by multiple rare variants with moderate to high penetrances • Associations through LD would not be suitable
Association tests for Common variants • Test a single marker each time • Cochran-Armitage’s trend test (CATT) (assuming additive (ADD)) • Power: High for additive (ADD) or Multiplicative (MUL); low recessive (REC) or Dominant (DOM) • Genotype association test (GAT) using chi-square statistic • Power: a little lower for ADD, higher for REC • MAX3 = maximum of three trend test statistics across the REC, ADD, and DOM models (Freidlin et al. 2002 Hum Hered.) • Power: lower than CATT under ADD • higher than CATT & CAT under REC
Association tests for Common variants • Test for single marker (CATT, GAT, & MAX3) • Low power when MAF <10% • No power for rare variants with MAF<1% • Multivariate test • Considering a group of variants (ex. SNPs in a gene) each time • Multiple logistic regression (or Hotelling test, Fisher’s product) • Xij = 0, 1, 2, the count. of the minor alleles of indivi at locus j • Power: higher than single-marker test; still very low due to large d.f = No. of SNPS = k • Need new methods for rare variants • Collapsing SNPs into a single marker to reduce d.f.
Outline • Introduction to association tests • Three well-known collapsing methods for rare variants: CAST, CMC, & Weighted Sum methods • An evaluation using GAW 17 data • Extension to the three collapsing methods • Future research
Three association tests for Rare variants • Collapsing a set of rare variants (into a single marker) • A cohort allelic sums test (CAST) (Morgenthaler & Thilly 2007, Mutat. Res.) • Combined Multivariate and Collapsing (CMC) (Li & Leal, 2007, AJHG) • Division into subgroups, collapsing in each subgroup • Weighted Sum statistic (Madsen & Browning, 2009; PloS Genet. Price et al. 2010, AJHG)
A cohort allelic sums test (CAST) • A group of n variants (SNPs) in a unit (ex. one gene, LD block) • Collapsing the genotypes across the variants • Indicator coding for individual j • xj = 1, if rare alleles present at any of the n variants; • xj= 0, otherwise • Testing if the proportions of individuals with rare variants (xj = 1) in cases and controls differ • Higher power than method testing single variant each time • Only for rare variants
Combined Multivariate and Collapsing (CMC) Method (Li & Leal 08) • Consider SNPs in a unit with MAF< a threshold (0.01 or 0.05) • Division and Collapsing • Divided into several sub-groups based on the MAF • Ex. Subgroups : (0, 0.001], [0.001, 0.005), [0.005, 0.01) • SNPs are collapsed in each sub-group • xij = 1, if indiv j has rare alleles present in the i-th subgroup; • xij= 0, otherwise
Combined Multivariate and Collapsing (CMC) Method (Li & Leal 08) • Multivariate test of collapsed sub-groups • Hotelling T2 test, MANOVA, Fisher’s product method • Power: often higher than CAST • Different threshold may have different power
Weighted Sum Method (Madsen & Browning 09) • A group of variants (SNPs) in a unit • A weight for SNP iby the S.t.d of No. of minor alleles in the sample • qiis the minor allele freq in controls • Calculate a weighted genetic score for indivj • Iij = 0, 1, 2, the count of the minor allele of indivi at locus j • Obtain the Rank (Vj); Sum of the ranks of affected indivs
Permutation for p-value estimation • From observed data: • Permutation to estimate p-value: • Phenotype labels are permuted 1000 times, x1, …x1000 • Calculate the mean (μ) and standard deviation (σ) of 1000 xs • Assume z ~ N(0, 1) under null hypothesis • Obtain the p-value from N(0, 1) • Fast, p-value ~U[0,1]
Weighted Sum Method (Madsen & Browning 09) • Power comparison: • Simulations assuming genotypic relative risk is proportion to MAF at disease loci (Madsen & Browning 09) • Weighted Sum Method (WSM) > CMC > CAST • (WSM) > CMC may not be true in other situations • Can be applied to rare variants & common variants • Disadvantage: • Give very high weights to very rare alleles (singleton), very low weights to common variants.
An evaluation of the CMC method and Weighted sum method by using GAW 17 data • Both methods are powerful (based on the authors’ simulation) • Our evaluation based on simulated datasets from GAW 17 • GAW 17 data: • a subset of genes with real sequence data available in the 1000 genome project • Simulated phenotypes • Unrelated individuals, families • Dataset of 697 unrelated individuals • 24487 SNPs in 3205 genes from 22 autosomal chromosomes • Only test for the 2196 genes with non-synonymous SNPs
GAW 17 dataset of unrelated individuals • Four phenotypes: Q1, Q2, Q4 and disease status. • Q1, Q2, and Q4 are quantitative traits • Q1 associated with 39 SNP in 9 genes, • Q2 associated with 72 SNPs in 13 genes • Q4: not related to any genes • Disease status is a binary trait: affected or unaffected, associated with 37 genes • 200 simulated phenotype replicates • Only one replicate of genotype data (original data)
Transforming Phenotypes • Methods: case-control design • Transform Q1, Q2, Q4 into binary traits • Splitting at the top 30% percentile of the distributions
Criteria for evaluation of Tests • Familywise error rate (FWER) • 2196 genes with non-synonymous SNPs, 2196 tests • 2196 null hypotheses Hj0: gene not associated with the trait • Q1 associated in 9 genes, 9 null hypotheses are not true. • (2196-9) null hypotheses are true • FWER = Pr(reject at least one true null hypothesis) = Nf/200 • Nf: No. of replicates, at least one true hypothesis are rejected • Average Power • Mean of power for all the 9 genes that affect the phenotypes • Evaluating power: Q1, Q2, Disease • Evaluate FWER: Q4
Distribution of MAF in the GAW 17 dataset Figure 1. Distribution of MAF of 24487 SNPs in GAW 17
Figure 1. Group SNPs based on MAFs for CMC Similar to Madsen & Browning (2009) 0 - 0.01 0.01 - 0.1 >=0.1
Table 2: FWER (nominal α = 0.05) • CMC has FWER inflation • Population stratification or admixture, • Samples from Asian, Europe,… • Relatedness among samples • Similar results in Power and FWER were reported at GAW 17
Variable-Threshold Approach (Price et al 2010) • Given a threshold T, calculate a score for indivj • Iij = 0, 1, 2, the count of the minor allele of indivi at locus j • Calculate the sum of score for cases: • Calculate Z(T) = V(T)/Var(V(T)) • Find T to maximize Z(T), Zmax = max (Z(T)) • Permutation to estimate p-value for Zmax • Power: >CMC; Extended to quantitative traits
A weighted approach (Price et al 2010) • Calculate a weighted score for indivj • Iij = 0, 1, 2 • Calculate the sum of score for cases • Possible weight • Power: similar to the weighted sum method (Madsen & Browning 09)
A weighted approach (Price et al 2010) • Calculate the sum of score for cases • Iij = 0, 1, 2 • Calculate weight by the prediction of functional effects • PolyPhen-2 is used to predict damaging effects of missense mutations with probabilistic scores. • Probabilistic scores as weights may reduce the noise of non-functional variants. • Higher Power than other methods
A data-adaptive sum test (Han & Pan 2010, Hum Hered) • Logistic model • xij = 0, 1, 2, the count of the minor allele of indivi at locus j • Effect on opposite directions • If j <0, with p-value < threshold (0.1), change xij into 2-xij • Permutation to estimate p-value
Conclusion • Collapsing methods have higher power than single-marker test • For genome-wide data analysis, collapsing methods don’t have much power after multiple testing adjusting • Weighted sum methods are promising, need prior information from biological data
Future research • Modifying the weighted sum method (in progress) • Very high weights to very rare variants • Smoothing weights w’ = 0.5w +0.5 (average of all w)