Download
1 / 18
180 likes | 187 Views
Explore protein sequence analysis tools such as protein modifications, sorting, and sub-cellular localization. Learn about algorithms like artificial neural networks and hidden Markov models for predicting protein characteristics.

E N D
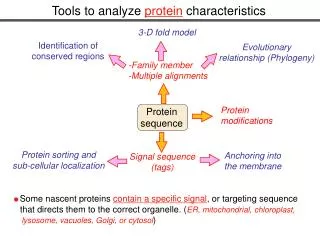
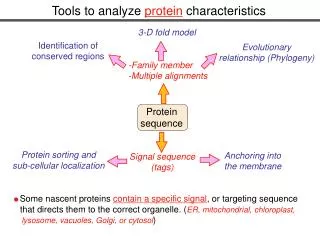
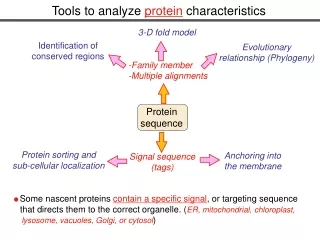
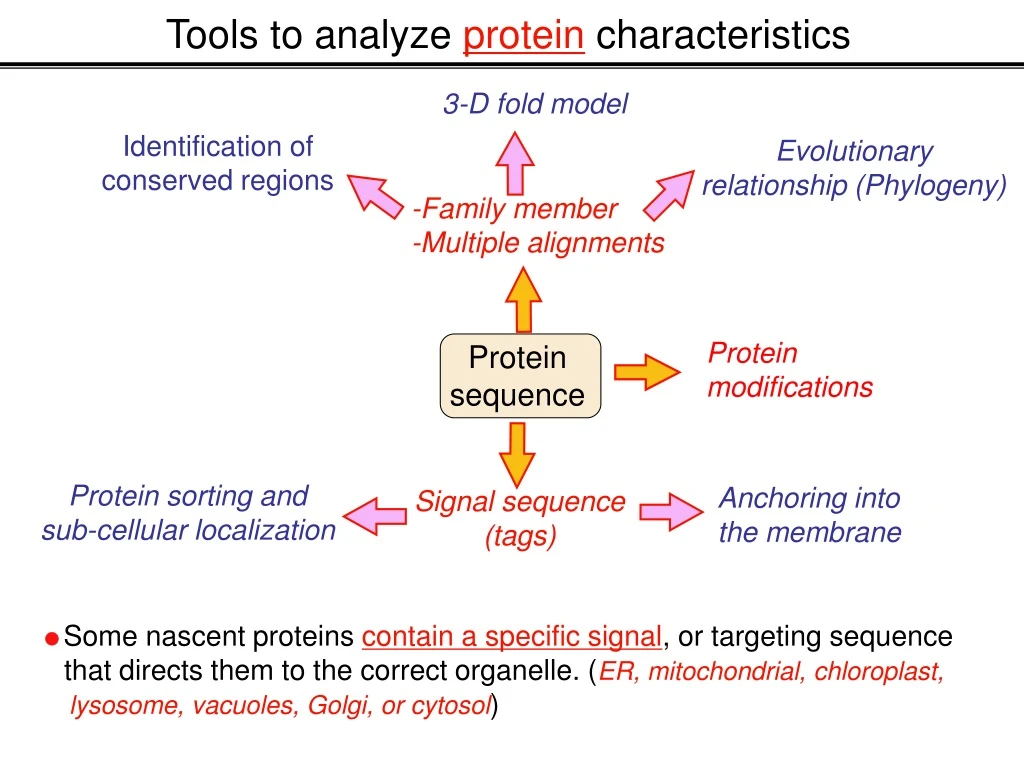
Protein sequence Protein modifications Protein sorting and sub-cellular localization Anchoring into the membrane Signal sequence (tags) Some nascent proteins contain a specific signal, or targeting sequence that directs them to the correct organelle. (ER, mitochondrial, chloroplast, lysosome, vacuoles, Golgi, or cytosol) Tools to analyze protein characteristics 3-D fold model Identification of conserved regions Evolutionary relationship (Phylogeny) -Family member -Multiple alignments
Learning algorithms are good for solving problems in pattern recognition because they can be trained on a sample data set. Classes of learning algorithms: -Artificial neural networks (ANNs) -Hidden Markov Models (HMM) Questions Can we train the computers: To detect signal sequences and predict protein destination? To identify conserved domains (or a pattern)in proteins? To predict the membrane-anchoring type of a protein? (Transmembrane domain, GPI anchor…) To predict the 3D structure of a protein?
Artificial neural networks (ANN) Machine learning algorithms that mimicthe brain. Real brains, however, are orders of magnitude more complex than any ANN. ANN is composed of a large number of highly interconnectedprocessing elements (neurons) working simultaneously to solve specific problems. ANNs, like people, learn by example. ANNs cannot be programmed to perform a specific task. The first artificial neuron was developed in 1943 by the neurophysiologist Warren McCulloch and the logician Walter Pits.
Hidden Markov Models (HMM) Used to answer questions like: What is the probability of obtaining a particular outcome? What is the best model from many combinations? HMM is a probabilisticprocess over a set of states, in which the states are “hidden”. It is only the outcome that visible to the observer. Hence, the name Hidden Markov Model. • HMM has many uses in genomics: • Gene prediction (GENSCAN) • SignalP • Finding periodic patterns
The ExPASy (Expert Protein Analysis System) Expasy server (http://au.expasy.org) is dedicated to the analysis of protein sequences and structures. Sequence analysis tools include: DNA -> Protein [Translate] Patternand profile searches Post-translational modification and topology prediction Primary structure analysis Structure prediction (2D and 3D) Alignment
PredictProtein:A service for sequence analysis, and structure prediction http://www.predictprotein.org/newwebsite/submit.html TMpred: http://www.ch.embnet.org/software/TMPRED_form.html TMHMM: Predicts transmembrane helices in proteins (CBS; Denmark) http://www.cbs.dtu.dk/services/TMHMM-2.0/ big-PI : Predicts GPI-anchor site:http://mendel.imp.univie.ac.at/sat/gpi/gpi_server.html DGPI: Predicts GPI-anchor site: http://129.194.185.165/dgpi/index_en.html SignalP: Predicts signal peptide: http://www.cbs.dtu.dk/services/SignalP/ PSORT: Predicts sub-cellular localization:http://www.psort.org/ TargetP: Predicts sub-cellular localization:http://www.cbs.dtu.dk/services/TargetP/ NetNGlyc: Predicts N-glycosylationsites:http://www.cbs.dtu.dk/services/NetNGlyc/ PTS1: Predicts peroxisomal targeting sequences http://mendel.imp.univie.ac.at/mendeljsp/sat/pts1/PTS1predictor.jsp MITOPROT: Predicts of mitochondrial targeting sequences http://ihg.gsf.de/ihg/mitoprot.html Hydrophobicity: http://www.vivo.colostate.edu/molkit/hydropathy/index.html
http://www.cbs.dtu.dk/services/: prediction server • NetNGlyc: Predicts N-glycosylation sites: http://www.cbs.dtu.dk/services/NetNGlyc/ • NetPhos: Predicts phosphorylation of residues: http://www.cbs.dtu.dk/services/NetPhos/ • NetPhosK: Predicts recognition sites for specific kinases: http://www.cbs.dtu.dk/services/NetPhosK/ • NetAcet: N-terminal acetylation in eukaryotic proteins: http://www.cbs.dtu.dk/services/NetAcet/ • NetCGlyc: C-mannosylation sites in mammalian proteins
Multiple alignment • Used to do phylogenetic analysis: • Same protein from different species • Evolutionary relationship: history • Used to find conserved regions • Local multiple alignment reveals conserved regions • Conserved regions usually are key functional regions • These regions are prime targets fordrug developments • Protein domains are often conserved across many species Algorithm for search of conserved regions: Block maker: http://blocks.fhcrc.org/blocks/make_blocks.html
Multiple alignment tools Free programs: Phylip and PAUP: http://evolution.genetics.washington.edu/phylip.html Phyml: http://atgc.lirmm.fr/phyml/ The most used websites : http://align.genome.jp/ http://prodes.toulouse.inra.fr/multalin/multalin.html http://www.ch.embnet.org/index.html(T-COFFEE and ClustalW) ClustalW: Standard popular software Italigns 2 and keep on adding a new sequence to the alignment Problem: It is simply a heuristics. Motif discovery: use your own motif to search databases: PatternFind: http://myhits.isb-sib.ch/cgi-bin/pattern_search http://meme.nbcr.net/meme4_6_0/intro.html
Phylogenetic analysis • Phylogenetic trees • Describe evolutionary relationships between sequences Major modes that drive the evolution: Point mutations modify existing sequences Duplications (re-use existing sequence) Rearrangement • Two most common methods • Maximum parsimony • Maximum likelihood The most useful software: http://www.megasoftware.net/mega4/m_con_select.html
Definitions Homologous:Have a common ancestor. Homology cannot be measured. Orthologous:The same gene in different species . It is the result of speciation (common ancestral) Paralogous: Related genes (already diverged) in the same species. It is the result of genomic rearrangements or duplication
Determining protein Structure-Function • Direct measurement of structure • X-ray crystallography • NMR spectroscopy Site-directed mutagenesis • Computer modeling • Prediction of structure • Comparative protein-structure modeling
Comparative protein-structure modeling Goal:Construct 3-D model of a protein of unknown structure (target), based on similarity of sequence to proteins of known structure (templates) • Procedure: • Template selection • Template–target alignment • Model building • Model evaluation Blue: predicted model by PROSPECT Red: NMR structure
The Protein 3-D Database The Protein DataBase (PDB) contains 3-D structural data for proteins Founded in 1971 with a dozen structures As of June 2004, there were 25,760 structures in the database. All structures are reviewed for accuracy and data uniformity. • 80% come from X-ray crystallography • 16% come from NMR • 2% come from theoretical modeling Structural data from the PDB can be freely accessed at http://www.rcsb.org/pdb/
Most used websites for 3-D structure prediction Protein Homology/analogY Recognition Engine (Phyre) at http://www.sbg.bio.ic.ac.uk/phyre/html/index.html PredictProtein at http://www.predictprotein.org/newwebsite/submit.html UCLA Fold Recognition at http://www.doe-mbi.ucla.edu/Services/FOLD/
Commercial bioinformatics softwares CLC Genomics Workbench Genomics: 454, Illumina Genome Analyzer and SOLiD sequencing data; De novo assembly of genomes of any size; Advanced visualization, scrolling, and zooming tools; SNP detection using advanced quality filtering; Transcriptomics: RNA-seq including paired data and transcript-level expression; Small RNA analysis; Expression profiling by tags; Epigenetics: Chromatin immunoprecipitation sequencing (ChIP-seq) analysis; Peak finding and peak refinement; Graph and table of background distribution; false discovery rate; Peak table and annotations; VectorNTI: Sequence analysis and illustration; restriction mapping; recombinant molecule design and cloning; in silico gel electrophoresis; synthetic biology workflows AlignX: BioAnnotator: ContigExpress: GenomBench
The bioinformatics not covered in this class Comparative genomics and Genome browser: http://genome.lbl.gov/vista/index.shtml http://www.sanger.ac.uk/resources/software/artemis/ Genome annotation: http://linux1.softberry.com/berry.phtml http:// rast.nmpdr.org/ Metagenomics: http://metagenomics.anl.gov/ System biology tools.