Download

1 / 24
240 likes | 357 Views
Predictive Analysis of Gene Expression Data from Human SAGE Libraries. Alexessander Alves* Nikolay Zagoruiko + Oleg Okun § Olga Kutnenko + Irina Borisova + * University of Porto, PORTUGAL + Russian Academy of Sciences RUSSIA

E N D
Predictive Analysis ofGene Expression Data fromHuman SAGE Libraries Alexessander Alves* Nikolay Zagoruiko+ Oleg Okun§ Olga Kutnenko+ Irina Borisova+ * University of Porto, PORTUGAL + Russian Academy of Sciences RUSSIA § University of Oulu FINLAND
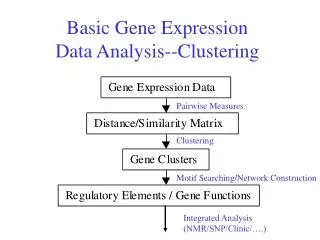
Outline • Goals • Background • SAGE Data • Gene Expression Data • Feature Selection • GRAD • Experiments • Conclusions
Goal • Predictive Analysis: • Feature Selection Methods in Bioinformatics and Machine Learning • Cancer Classification
Background Central Dogma of Biology • Genes code proteins and other larger biomolecules • Genes are expressed in a two steps process (Central Dogma of Biology) • Several technologies measure transcription: SAGE, Micro array… Molla et al, 2003 Gene Expression Process 1- Transcribed into an RNA Sequence 2- Translated into a protein
SAGE DATA • Advantages: • Compare samples between different organs and patients. (No normalisation required) • Collects complete gene expression profile of a cell/tissue without prior knowledge of the mRNA to be profiled
SAGE DATA • Drawbacks: • Very Expensive to Collect Data using the SAGE method • Very Few Examples (consequence)
GENE EXPRESSION DATA • Challenges posed to Machine Learning • Number of Genes Dramatically Exceeds Examples!!! • Curse of Dimensionality (not enough density to estimate accuratelly the model) • Over-fitting (higher probability of finding casual relationships among data attributes)
Feature Selection • Remove Irrelevant and Redundant Genes • Methods: • Wrapper • Fit classifier to a subset of data and use classification accuracy to drive the search for relevant genes (e.g. C4.5 accuracy ) • Filtering • Use a function to assess the goodness of a subset of genes (e.g. euclidean distance, entropy, correlation, etc...) • Problem Complexity • O(2n) ... • n, number of genes • Smaller dataset n=822. • O(2n) 2.8x10246 Intractable using a simple exaustive search
Gene Selection In Bioinformatics • Filtering is usually prefered because is computationally less expensive • Several works on classification select genes with: • Wilcoxon test, • t-test • Additionally, also remove genes with low entropy, variability, or absolute expression level. • Cons • Redundancy • Interdependency unaware
Our Proposals • Study Bioinformatics Filtering Techniques • Compare with Machine Learning Algorithms • Avoid Redundancy • Consider Interdependency and low expressed genes • Introduce a new Filtering Algorithm GRAD
GRAD • Search Strategy • Use Exaustive Searchon the formation of informative groups of attributes (“granules”) • Use AdDelfor choosing subsets of granules • AdDel: A combination of forward sequential search (FSS) and backward sequential search (BSS) • Number of attributes to include on a subset is estimated by algorithm
GRAD • Algorithm P0: x1,x2,…,xn – initial set of features Formation of granules: Ordering by individual relevance G1: x7, x33, x12,…,xn All pairs by exhaustive search G2: x3x8, x15x88,…,xi xj All triplets by exhaustive search G3: x75x1x35, x11x49x55,…, xi xj xk Top level most relevant granules using AdDel • G=<G1,G2,G3>… AdDel and are the distances to closest neighbors, one from each class
Experiments • Comparison • GRAD • Wrapper C4.5 • Original Dataset • Filtering • Wilcoxon Test, low entropy, variability, and very low absolute expression level • Classifiers • C4.5 • SVM • RBF • NN-MLP • Data • Small Dataset:74x822
Data Characterization • Not all organs have samples of both classes • Unbalanced number of cases: • 50 Cancer Samples • 24 Normal Samples Most data is relativelly low expressed Mean quite far from median: Potentially due to outliers
Data Characterization average vs standard deviation average vs range Both range and standard deviation have roughly linear relationship with gene expression level average
Experimental Results Predictive Accuracy GRAD WRAPPER Original Filtering 86% 82% 79% 78% GRAD is significantly better than using the original or the filtered dataset Wrapper approach is not
GRAD Results • Importance of considering dependence • Distance Function: • best by GRAD • P=100 % 10 most individually informative P=75,7 %
GRAD Results • Scatter Plot of GRAD Attributes Interdependency relationship between two non differentially expressed genes selected with GRAD Two differentially expressed genes selected with GRAD.
GRAD Results • Examples ordered by the value of the Distance Function In the future it can allow to estimate the degree of risk, to make early diagnostics and to supervise a course of treatment
Induced Classifiers C4.5 Induced on GRAD attributes C4.5 Induced using a Wrapper Approach
Conclusions • Coping with redundancy and dependency between attributes is very important. • Algorithm GRAD represents effective meansto select a subset of attributes from very big initial set. • The submitted results have only illustrative character. • We are open for cooperation with those who have interest on the biological interpretation of results
Questions • …
GRAD • In increasing n the relevance grows, then growth stops and begins its decrease due to addition less informative, rustling attributes. • The maximum of the curve of quality allows • to specify optimum quantity of attributes. • Only algorithms of AdDel family has such property.
Feature Selection • Wrapper • Considers the classifier while searching best subset • Accuracy Improves • May overfit due to small sample sizes and huge dimensionality • Computationally more expensive • Filtering: • Potentially less accurate • Faster: Does not requires the induction of a predictor • Commonly prefered approach in bioinformatics