Download

1 / 62
620 likes | 761 Views
Inferring Protein Structure with Discriminative Learning and Network Diffusion. Rui Kuang Department of Computer Science Center for Computational Learning Systems Columbia University Thesis defense, August 16 th 2006. Agenda. Biological Background

E N D
Inferring Protein Structure with Discriminative Learning and Network Diffusion Rui Kuang Department of Computer Science Center for Computational Learning Systems Columbia University Thesis defense, August 16th 2006
Agenda • Biological Background • Protein Classification with String Kernels • Domain Identification and Boundary Detection • Protein Ranking with Network Diffusion • Conserved Motifs between Remote Homologs • Conclusion & Future Research
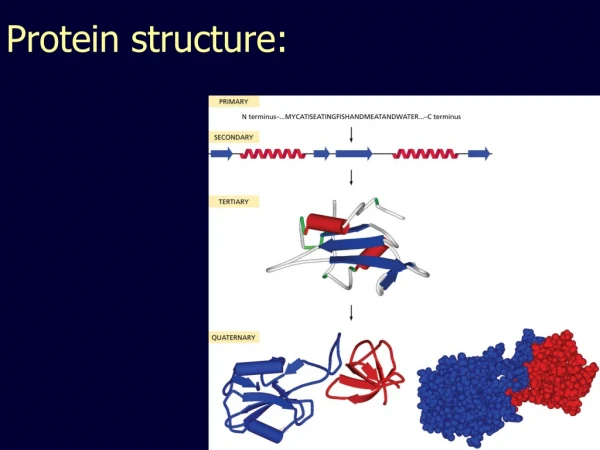
Background Structural classification Domain segmentation Protein ranking Motif discovery Conclusion & Future Research What are proteins? Proteins – encoded by genes • A protein (polypeptide chain) is a sequence of amino acid residues • Derived from Greek word proteios meaning “of the first rank” by Jöns J. Berzelius in 1838 Amino Acid Polypeptide Chain (Picture courtesy of Branden & Tooze )
Why study proteins? • Proteins play crucial functional roles in all biological processes: enzymatic catalysis, signaling messengers, structural elements… • Function depends on unique 3-D structure. • Easy to obtain protein sequences, difficult to determine structure. NLAFALSELDRITAQLKLPRHVEEEAARLYREAVRKGLIRGRSIESVMAACVYAACRLLKVPRTLDEIADIARVDKKEIGRSYRFIARNLNLTPKKLF… fold determine Sequence Structure Function (Picture courtesy of Branden & Tooze )
Sequence Space and Structure Space Sequence (>2,500,000) • Homologous proteins share >30% sequence identity, which suggests strong structural similarity. • Remote homologous proteins share similar structure but low sequence similarity. Structure (38,086 known in PDB): discrete groups of folds with unclear boundaries (by 8/14/2006)
Remote Homology Detection • Remote homology: remote evolutionary relationship conserved structure/function, low sequence similarity • It is often not possible to detect statistically significant sequence alignment between remote homologs <10% sequence identity ADTIVAVELDTYPNTDIGDPSYPHIGIDIKSVRSKKTAKWNMQNGKVGTAHIIYNSVDKRLSAVVSYPNADSATVSYDVDLDNVLPEWVRVGLSASTGLYKETNTILSWSFTSKLKSNSTHETNALHFMFNQFSKDQKDLILQGDATTGTDGNLELTRVSSNGPQGSSVGRALFYAPVHIWESSAVVASFEATFTFLIKSPDSHPADGIAFFISNIDSSIPSGSTGRLLGLFPDAN MSLLPVPYTEAASLSTGSTVTIKGRPLVCFLNEPYLQVDFHTEMKEESDIVFHFQVCFGRRVVMNSREYGAWKQQVESKNMPFQDGQEFLSISVLPDKYQVMVNGQSSYTFDHRIKPEAVKMVQVWRDISLTKFNVSYLKR DSSYYWEIEASEVMLSTRIGSGSFGTVYKGKWHG-DVAVKILKVVDPTPEQFQAFRNEVA D + WEI+ +++ + ++ SGS+G +++G ++VA+K LK E +F EV DGTDEWEIDVTQLKIEKKVASGSYGDLHRGTYCSQEVAIKFLKPDRVNNEMLREFSQEVF
Protein Domains • Proteins often consist of several independent domains • fold autonomically • often function differently • represent fundamental structural, functional and evolutionary units • Example: a two-domain protein • 3-layer(aba) sandwich at the N-terminal • a mainly alpha in an orthogonal bundle at the C-terminal
Boundary identification Domain 1 Domain 2 Domain 3 Remote homology detection Inferring protein structure/function from sequence similarity • For newly sequenced genomes, often homology detection can only identify less than a half of the genes. • Remote homology detection and domain segmentation are crucial steps for studying genes with no close homology. Query sequence Sequence Alignment Domain Family 1 Domain Family 2 Domain Family 3 Domain Family n-2 Domain Family n-1 We want to correctly segment protein sequences into domains (domain boundary identification) and associate them with their corresponding structural/functional class (remote homology detection). Domain Family n Domain database
Background Structural classification Domain segmentation Protein ranking Motif discovery Conclusion & Future Research Superfamily : low sequence similarity but functional features suggest probable common evolutionary origin Family : Sequence identity > 30% or functions and structures are very similar Protein Structural Classification • Protein classification is the prediction of the structural or functional class of a protein from its primary sequence. • SCOP: Structural Classification of Proteins • Known domain structures are organized in a hierarchy: family, superfamily and fold. SCOP
Negative Test Set Negative Training Set Positive Training Set Positive Test Set Remote Homology Detection in Protein Classification • Remote homologs: sequences that belong to the same super-family but not the same family. SCOP Fold Super-family Family
Support Vector Machine (SVM) Classifiers • SVM: Large margin-based discriminative learning approach. • Find a hyperplane to separate positive data from negative data and also maximize the margin. • A Quadratic Programming problem only depends on the inner product between data points.
Kernels for Discrete Objects • Kernel trick: To train an SVM, can use kernelrather than explicit feature map • Can define kernels for sequences, graphs, other discreteobjects: { sequences } RN Kernel value is inner product in feature space: K(x, y) = (x), (y) • Original string kernels (Watkins, Haussler, Lodhi et al.) require quadratic time in sequence length, O(|x| |y|), to compute each kernel value K(x, y). • We introduce fast novel string kernels with linear time complexity.
Profile Kernel and its Family Tree • Three generations • Spectrum Kernels • Mismatch Kernels • Profile Kernels • Faster computation, i.e., linear computation time in sequence length. • Profile-based string kernels take advantage of abundant unlabeled data to capture homologous/evolutionary information for remote homology detection.
K-mers capture some position-independent local similarity, but they don’t effectively model evolutionary divergence. Spectrum Kernel Feature map indexed by all possible k-length subsequences (“k-mers”) from alphabet of amino acids, || = 20 Feature Space (AAA-YYY) 1 AKQ 1 1 DYY 1 0 EIA 1 0 IAK 1 1 KQD 0 0 KQE 1 1 QDY 0 0 YEI 1 1 YYE 1 2 YYY 0 Q2:DYYEIAKQE Q1:AKQDYYYYE DYY YYE YEI EIA IAK AKQ KQE AKQ KQD QDY DYY YYY YYY YYE K(Q1,Q2)= <(…1…1…0…0…1…0…1…0…1…2),(…1…1…1…1…0…1…0…1…1…0)> =3 Leslie, Eskin and Noble, PSB 2002
Mismatch Kernel • For k-mer s, the mismatch neighborhood N(k,m)(s) is the set of all k-mers t within m mismatches from s • Size of mismatch neighborhood is O(||mkm) AKQ … CKQ AKY … DKQ AAQ ( 0 , … , 1 , … , 1 , … , 1 , … , 1 , … , 0 ) AAQ AKY CKQ DKQ AKQ Arbitrary mismatch does not model the mutation probability between amino acids. Leslie, Eskin, Weston and Noble, NIPS 2002
Profile Kernel [Kuang & Leslie, JBCB 2005 and CSB 2004] • Profile kernel: specialized to protein sequences, probabilistic profiles to capture homology information • Semi-supervised approach: profiles are estimated using unlabeled data (about 2.5 million proteins ) • E.g. PSI-BLAST profiles: estimated by iteratively aligning database homologs to query sequence. • Profiles are build from multiple sequence alignment to model the positional mutation probability. L K L … A 3 -2 1 … C -1 0 2 … D -1 0 0 … … … … … … Y 2 -3 -3 … QUERYLKLLRFLGSGAFGEVYEGQLKTE....DSEEPQRVAIKSLRK....... HOMOLOG1IIMHNKLGGGQYGDVYEGYWK........RHDCTIAVKALK........ HOMOLOG2LTLGKPLGEGCFGQVVMAEAVGIDK.DKPKEAVTVAVKMLKDD...... HOMOLOG3 IVLKWELGEGAFGKVFLAECHNLL...PEQDKMLVAVKALK........
Profile-based k-mer Map • Use profile to define position-dependent mutation neighborhoods: • E.g. k=3, =5 and a profile of negative log probabilities A K Q … A 1 3 4 … C 5 4 1 … D 4 4 4 … … … … … … K 4 1 4 … … … … … … Q 3 4 1 … … … … … … Y 2 4 3 … AKQ YKQ (2+1+1<) YKC (2+1+1<) AKQ (1+1+1<) AKC (1+1+2<) ( 0 , … , 1 , … , 1 , … , 1 , … , 1 , … , 0 ) AKC AKQ YKC YKQ AKQ
Efficient Computing with Trie • Use trie data structure to organize lexical traversal of all instances of k-mers in the training profiles. • Scales linearly with length, O(km_max+1||m_max(|x|+|y|)), where m_max is maximum number of mismatches that occur in any mutation neighborhood. • E.g. k=3, =5 Sequence y Sequence x … A Q Y … A ….5 2 1 … C … 2 1 2 … D … 2 1 4 … … … … … … … Q … 2 .6 2 … … … … … … … Y … 3 3 3 … A Q K … A 1 3 2 … C 3 2 1 … D 3 2 1 … … … … … … Q 3 1 2 … … … … … … Y 2 1 3 … A Q C D x: 1+1+1 < y: .5+.6+4 > x: 1+1+1< y: .5+.6+2 < Update K(x, y) by adding contribution for feature AQC but not AQD
Inexact Matching Kernels[Leslie & Kuang, JLMR 2004, KMCB 2004 & COLT 2003] • Gappy kernels • For g-mer s, g > k, the gapped match set G(g,k)(s) consists of all k-mers t that occur in s with up to (g - k) gaps • Wildcard kernels • Introduce wildcard character “”, define feature space indexed by k-mers from {}, allowing up to m wildcards • Substitution kernels • Use substitution matrices to obtain P(a|b), substitution probabilities for residues a, b • The mutation neighborhood M(k,)(s) is the set of all k-mers t such that - i=1…k log P(si|ti) <
Experiments • SCOP 1.59 benchmark with 54 experiments • Train PSI-BLAST profiles on NR database • Comparison against PSI-BLAST and recent SVM-based methods: • PSI-BLAST rank: use training sequence as query and rank testing sequences with PSI-BLAST e-value • eMotif Kernel (Ben-Hur et al., 2003): features are known protein motifs, stored using trie • SVM-pairwise (Liao & Noble, 2002): feature vectors of pairwise alignment scores (e.g. PSI-BLAST scores) • Cluster Kernel (Weston et al., 2003): Implicitly average the feature vectors for sequences in the PSI-BLAST neighborhood of input sequence
Background Structural classification Domain segmentation Protein ranking Motif discovery Conclusion & Future Research Boundaries Identify Protein Domains and Domain Boundaries • SVM-based remote homology detection methods do not rely on sequence alignment. • To learn the domain segmentation, we use our SVM classifiers as domain recognizers and find the optimal segmentation giving the maximum sum of the classification scores. SCOP Super-families: SVM2 SVM3 SVM4 SVM1 Domain recognizers: Query sequence
Algorithms for finding optimal segmentation • Assuming we know the number of domains on a protein, we look for the optimal segmentation with the maximum sum of classification scores with dynamic programming. • No gaps: • Allowing gaps (can also be solved as a LP problem): : segment between position i and position j of sequence S : the best classification score of segment s : the maximum sum of classification scores from c segments on S1,j
Toy Example of Dynamic Programming 1 4 2 2 1 1 c=1 -INF 2 5 8 6 7 c=2 c=3 -INF -INF 3 6 9 13
boundary region domain region Algorithms for finding optimal segmentation (unconstrained number of domains) • The regions across boundaries are less classifiable than other regions within one domain. • Use dynamic programming to alternate between domain regions and boundary regions. : segment between position i and position j of sequence S : confidence score of assigning s as domain region : confidence score of assigning s as boundary region
Experiments • Datasets: • 25 SCOP folds: 2678 training domains and 471 test chains (189 multi-domain proteins). • 40 SCOP super-families: 1917 training domains and 375 test chains (131 multi-domain proteins). • Baseline approach: • Align test proteins against PSI-BLAST profiles of the training domains, and use the best aligned regions as domain regions. • Evaluation: • Domain label: significant overlap between the true domain and the predicted domain. • Boundaries: both the predicted start and end positions should be close to the true ones.
Experiments (Cont.) At least 75% percent positional overlap between the true domain and the prediction.
Background Structural classification Domain segmentation Protein ranking Motif discovery Conclusion & Future Research Protein Ranking • Protein ranking: search protein database for sequences that share an evolutionary or functional relationship with a given query sequence. • Standard protein ranking algorithm: pairwise alignment-based algorithm, PSI-BLAST, can easily detect close homologs. • Pairwise alignment-based algorithms are not effective for remote homolog detection. Query Homologous protein Remote homolog Other labeled protein
Cluster assumption: proteins with structural or functional relation tend to be in the same cluster in the network. Diffusion on the protein similarity network to capture cluster structure. Correct Ranking 1 2 3 1 2 3 4 5 6 7 8 6 7 8 4 5 Ranking based on local similarity From Local Similarity to Global Structure (RankProp) Weston, Elisseff, Zhou, Leslie, Noble, PNAS 2004 Noble, Kuang, Leslie, Weston, FEBS 2005 Weston, Kuang, Leslie, Noble, BMC Bioinformatics 2005 6 2 5 3 7 Query 4 1 Homologous protein Remote homolog Other labeled protein Unlabeled protein 8
RankProp (Cont.) • Capture global structure with diffusion. • Protein similarity network: • Graph nodes: protein sequences in the database • Directed edges: weighted by PSI-BLAST e-value • Initial ranking score at each node: the similarity to the query sequence • Iterative diffusion operation: : Initial ranking score : Ranking score at step t K : Normalized connectivity matrix : a parameter for balancing the initial ranking score and propagation
Query MotifProp [Kuang, Weston, Noble, Leslie Bioinformatics, 2005] • Motivated by HITS algorithm for page ranking and NLP algorithms • Protein-motif network • Nodes: proteins and motifs • Edges: whether a motif is contained in a protein • Motifs: patterns/models built on protein segments conserved during evolution. • Often characterize structural/ function properties of a protein. • Examples:eMOTIF, PROSITE, K-mers, BLOCKS… …FYPGKGHTEDNIVVWLPQYNILVGGCLVKSTSAKDLGNVADAYVNEWSTSIENVLKRYRNINAVVPGHGEVG… Motif Database
In MotifProp, protein nodes and motif nodes enforce their similarity to the query sequence through propagation. MotifProp (Cont.) • MotifProp can identify motif-rich regions derived from motif ranking to help interpret diffusion algorithm. • Low computational cost: protein-motif network is fast to build. • Motifs serve as bridges connecting homologous/remote homologous proteins. Query Protein vertices Motif vertices
Query Protein vertices Motif vertices Diffusion in Protein-motif Network MotifProp: Normalize affinity matrix H to Initialize P and F with the initial activation value Iterate until converge ( ) For all For all
Experiments • 7329 sequences (4246 for training and 3083 for testing) of <95% identity from SCOP 1.59 plus 100,000 proteins from Swiss-Prot. • Motif sets: 4-mers, PROSITE and eMOTIF.
Background Structural classification Domain segmentation Protein ranking Motif discovery Conclusion & Future Research Conserved Motifs between Remote Homologs • We can derive weights of k-mer features from SVM classifiers trained with profile kernel. • MotifProp provides activation values on the k-mer features after propagation. • Both the SVM weights and Motif activation values can be mapped back to protein sequences to identify conserved structural/functional motifs. • Positional contribution to classification score: where Δ is the SVM weights or MotifProp activation values on k-mer features.
Mapping Discriminative Regions to Structure (Profile Kernel) • In examined examples, discriminative motif regions correspond to conserved structural features of the protein superfamily • Example: Homeodomain-like protein superfamily. Ecoli MarA protein (1bl0)
Motif Rich Regions (MotifProp) • Motif-rich regions on chain B of arsenite oxidase protein from the ISP protein super-family. • The PDB annotation and motif-rich regions are given. • The 3D protein structure with motif-rich regions in yellow.
Background Structural classification Domain segmentation Protein ranking Motif discovery & angle prediction Conclusion & Future Research Conclusions &Contributions • Profile-based string kernels exploit compact representation of homology information for better detection of remote homologs. • Dynamic Programming-based approach improves multi-label domain classification and domain boundary detection over PSI-BLAST alignment-based approach. • MotifProp improves protein ranking over PSI-BLAST by network diffusion on protein-motif network. • Interpretation of profile-SVM classifier and MotifProp by motif regions: conserved structural components. • Fast kernels for inexact string matching. • Classifiers for protein backbone angle prediction (not presented).
Future Research • Protein function inference by structural genomics and proteomics • Identify functional properties of protein structures with kernel methods, e.g. prediction of protein functional sites and structure-based identification of protein-protein interaction sites. • Protein function inference from proteomics, e.g. protein function prediction based on protein-protein interaction patterns and protein structures. • Protein structure prediction • Unified prediction of protein backbone and side chain positions (Phi-Psi angles and rotamers) with energy-based cost function.
Acknowledgements: Committee • Tony Jebara Dept. of Computer Science, Columbia University • Christina Leslie (advisor) Center for Computational Learning Systems & C2B2, Columbia University • Kathleen Mckeown (chair) Dept. of Computer Science, Columbia University • William Stafford Noble Dept. of Genome Science & Dept. of Computer Science, University of Washington • Rocco Servedio Dept. of Computer Science, Columbia University • Jason Weston Machine Learning Group, NEC Labs (USA)
Acknowledgements: Collaborators • An-Suei Yang (Genome Research Center, Academia Sinica of Taiwan) • Dengyong Zhou (Machine Learning Group, Microsoft) • Yoav Freund (Computer Science Department, UCSD) • Eugene Ie (Computer Science Department, UCSD) • Ke Wang (Computer Science Department, Columbia University) • Wei Chu (Center For Computational Learning Systems, Columbia University) • Kai Wang (Biomedical Informatics Department, Columbia University) • Iain Melvin (Machine Learning Group, NEC) • Girish Yao (Computer Science Department, Columbia University) • Lan Xu (Molecular Biology Department, The Scripps Research Institute)
Publications • Structural classification: • Profile kernels (JBCB 2005 and CSB 2004) • Inexact marching kernels (JMLR 2004, COLT 2003 & KMCB 2004) • Protein ranking: • RankProp (FEBS 2005 and BMC Bioinformatics 2005) • MotifProp (Bioinformatics 2005) • Protein local structure prediction • Kernel methods based on sliding-window (Bioinformatics 2004) • Structured output learning (ongoing research) • Protein domain segmentation(In preparation)
Protein backbone angle prediction Conformational States A A A G B B B B B … Phi-Psi Angles …… (Φ1,Ψ1) (Φ2,Ψ2) (Φ3,Ψ3) (Φ4,Ψ4) (Φ5,Ψ5) (Φ6,Ψ6) (Φ7,Ψ7) (Φ8,Ψ8) …… 3-D structure Discretization of Phi-Psi angles
Sliding-window SVM approach [Kuang, Leslie & Yang 2004] Encode each position independently with sequence information within a length-k window. Conformational States A A A B B B B G G E B B B B B A:-3 –4 –4 –4 –3 –4….. A:0 –1 –1 3 –4 3 4 1….. B:0 –1 2 1 –3 4 0 –1…… B:-2 –3 –4 –5 –2 4…… B:0 –3 –1 –2 –4 –1…… …… To SVM Smoothing: use predictions to train a second sets of SVMs
Experiments • Datasets: 697 sequences of 97,365 amino acids with sequence identity < 25 % from PDB (PDB_SELECT25). • Comparison against: • LSBSP1: query against local structure-based sequence profile database. • HMMSTR: Hidden Markov Model based on local structural motifs.
Regularization Framework Closed form solution of MotifProp Related to the regularization framework in Zhou et. al. NIPS 2003 , Where • Initial Ranking : Final Ranking : • Normalized Affinity Matrix :