Download
1 / 35
350 likes | 731 Views
Modelling Proteomes Ram Samudrala University of Washington. Rationale for understanding protein structure and function. structure determination structure prediction. Protein structure - three dimensional - complicated - mediates function. homology rational mutagenesis

E N D
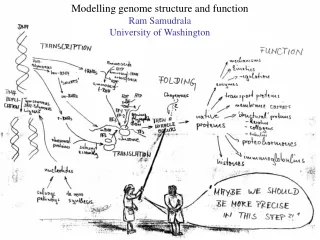
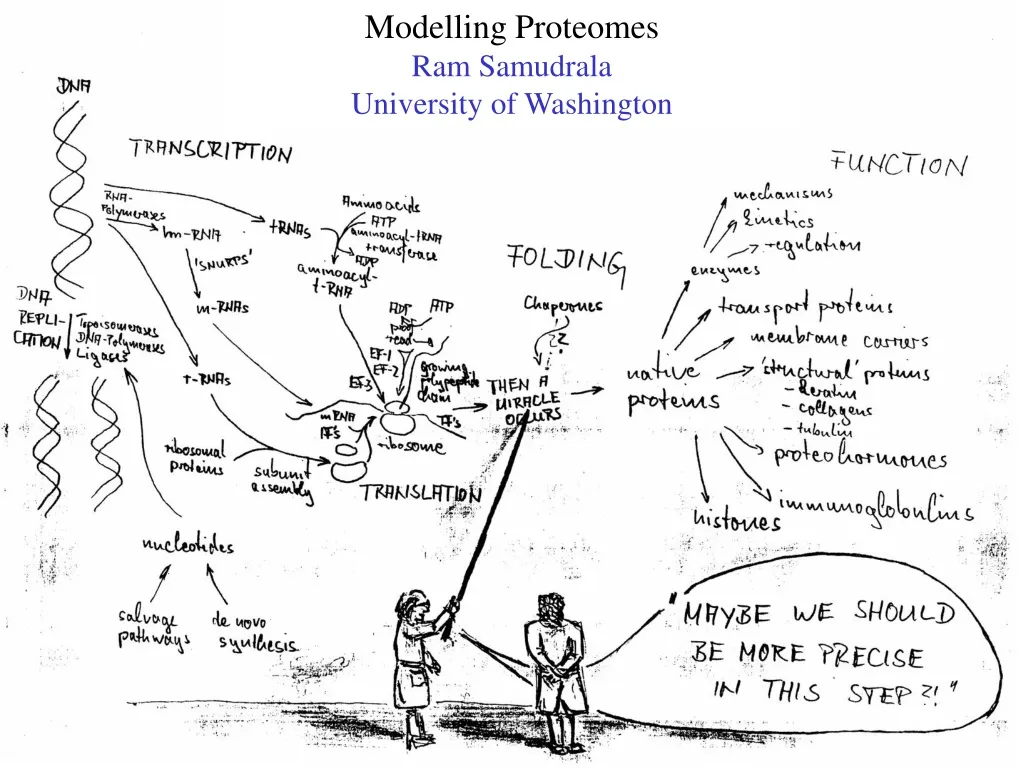
Modelling Proteomes Ram Samudrala University of Washington
Rationale for understanding protein structure and function structure determination structure prediction Protein structure - three dimensional - complicated - mediates function homology rational mutagenesis biochemical analysis model studies Protein sequence -large numbers of sequences, including whole genomes ? Protein function - rational drug design and treatment of disease - protein and genetic engineering - build networks to model cellular pathways - study organismal function and evolution
Protein folding not unique mobile inactive expanded irregular spontaneous self-organisation (~1 second) native state DNA …-CUA-AAA-GAA-GGU-GUU-AGC-AAG-GUU-… protein sequence …-L-K-E-G-V-S-K-D-… one amino acid unfolded protein
Protein folding not unique mobile inactive expanded irregular spontaneous self-organisation (~1 second) unique shape precisely ordered stable/functional globular/compact helices and sheets native state DNA …-CUA-AAA-GAA-GGU-GUU-AGC-AAG-GUU-… protein sequence …-L-K-E-G-V-S-K-D-… one amino acid unfolded protein
De novo prediction of protein structure select sample conformational space such that native-like conformations are found hard to design functions that are not fooled by non-native conformations (“decoys”) astronomically large number of conformations 5 states/100 residues = 5100 = 1070
Semi-exhaustive segment-based folding fragments from database 14-state f,y model generate … … monte carlo with simulated annealing conformational space annealing, GA minimise … … all-atom pairwise interactions, bad contacts compactness, secondary structure filter EFDVILKAAGANKVAVIKAVRGATGLGLKEAKDLVESAPAALKEGVSKDDAEALKKALEEAGAEVEVK
Decomposition of all-atom function using ICA (blind separation of sources by maximising the statistical independence across various channels) atom type 2 atom type 2 energy energy distance (A) distance (A) atom type 1 atom type 1 atom type 2 atom type 2 energy energy distance (A) atom type 1 atom type 1 distance (A) Disulphide bridges Main chain hydrogen bonding Salt bridges Side -> main chain hydrogen bonding Shing-Chung Ngan
Ab initio prediction at CASP Before CASP (BC): “solved” (biased results) CASP2: worse than random with one exception CASP1: worse than random CASP3: consistently predicted correct topology - ~ 6.0 Å for 60+ residues CASP4: consistently predicted correct topology - ~4-6.0 A for 60-80+ residues *T98/sp0a – 6.0 Å (60 residues; 37-105) **T102/as48 – 5.3 Å (70 residues; 1-70) **T97/er29 – 6.0 Å (80 residues; 18-97) **T106/sfrp3 – 6.2 Å (70 residues; 6-75) **T110/rbfa – 4.0 Å (80 residues; 1-80) *T114/afp1 – 6.5 Å (45 residues; 36-80)
Prediction for CASP4 target T110/rbfa Ca RMSD of 4.0 Å for 80 residues (1-80)
Prediction for CASP4 target T97/er29 Ca RMSD of 6.2 Å for 80 residues (18-97)
Prediction for CASP4 target T106/sfrp3 Ca RMSD of 6.2 Å for 70 residues (6-75)
Prediction for CASP4 target T98/sp0a Ca RMSD of 6.0 Å for 60 residues (37-105)
Prediction for CASP4 target T126/omp Ca RMSD of 6.5 Å for 60 residues (87-146)
Prediction for CASP4 target T114/afp1 Ca RMSD of 6.5 Å for 45 residues (36-80)
Postdiction for CASP4 target T102/as48 Ca RMSD of 5.3 Å for 70 residues (1-70)
Comparative modelling of protein structure scan align KDHPFGFAVPTKNPDGTMNLMNWECAIP KDPPAGIGAPQDN----QNIMLWNAVIP ** * * * * * * * ** … … build initial model construct non-conserved side chains and main chains minimum perturbation graph theory, semfold refine physical functions de novo simulation
A graph theoretic representation of protein structure -0.6 (V1) represent residues as nodes -0.5 (I) -0.9 (V2) weigh nodes -0.7 (K) -1.0 (F) construct graph -0.6 (V1) -0.2 -0.5 (I) -0.9 (V2) -0.1 -0.5 (I) -0.9 (V2) -0.1 -0.1 -0.3 -0.1 find cliques -0.2 -0.4 -0.3 -0.1 -0.1 -0.4 W = -4.5 -0.2 -0.7 (K) -1.0 (F) -0.2 -0.7 (K) -1.0 (F)
Comparative modelling at CASP alignment side chain short loops longer loops BC excellent ~ 80% 1.0 Å 2.0 Å CASP1 poor ~ 50% ~ 3.0 Å > 5.0 Å CASP2 fair ~ 75% ~ 1.0 Å ~ 3.0 Å CASP3 fair ~75% ~ 1.0 Å ~ 2.5 Å CASP4 fair ~75% ~ 1.0 Å ~ 2.0 Å CASP4: overall model accuracy ranging from 1 Å to 6 Å for 50-10% sequence identity **T111/eno – 1.7 Å (430 residues; 51%) **T122/trpa – 2.9 Å (241 residues; 33%) **T128/sodm – 1.0 Å (198 residues; 50%) **T125/sp18 – 4.4 Å (137 residues; 24%) **T112/dhso – 4.9 Å (348 residues; 24%) **T92/yeco – 5.6 Å (104 residues; 12%)
Prediction for CASP4 target T128/sodm Ca RMSD of 1.0 Å for 198 residues (PID 50%)
Prediction for CASP4 target T111/eno Ca RMSD of 1.7 Å for 430 residues (PID 51%)
Prediction for CASP4 target T122/trpa Ca RMSD of 2.9 Å for 241 residues (PID 33%)
Prediction for CASP4 target T125/sp18 Ca RMSD of 4.4 Å for 137 residues (PID 24%)
Prediction for CASP4 target T112/dhso Ca RMSD of 4.9 Å for 348 residues (PID 24%)
Prediction for CASP4 target T92/yeco Ca RMSD of 5.6 Å for 104 residues (PID 12%)
Protein structure from combining theory and experiment Ling-Hong Hung
Prediction of HIV-1 protease-inhibitor binding energies with MD 1.0 0.5 with MD without MD Correlation coefficient ps 0 0.2 0.4 0.6 0.8 1.0 MD simulation time Ekachai Jenwitheesuk
Bioverse – explore relationships among molecules and systems http://bioverse.compbio.washington.edu Jason Mcdermott
Bioverse – explore relationships among molecules and systems Jason Mcdermott
Bioverse – human protein-protein interaction network Jason Mcdermott/Zach Frazier
Bioverse – salmonella protein-protein interaction network Jason Mcdermott/Zach Frazier
Bioverse – human protein-protein similarity network Jason Mcdermott/Zach Frazier
Take home message Acknowledgements Ekachai Jenwitheesuk Jason McDermott Ling-Hong Hung Shing-Chung Ngan Yi-Ling Chen Zach Frazier Group members Levitt and Moult groups Prediction of protein structure and function can be used to model whole proteomes to understand organismal function and evolution