Download
1 / 45
450 likes | 576 Views
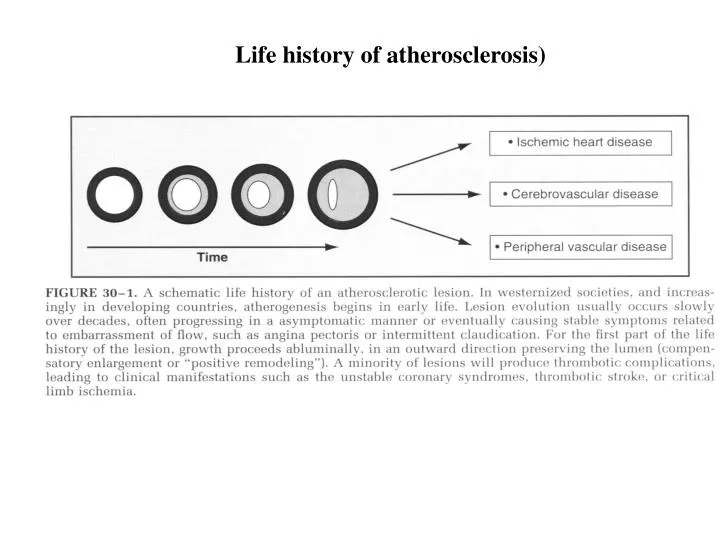
Life history of atherosclerosis). Structure of normal muscular artery. Evolution of atherosclerosis. VGH. 1. Modification of LDL ( ox-LDL) in the intima proteoglycan 2. Accumulation of LDL from oxidative stress 3. Elaboration of cytokines (interleukin-1, IL-1 )

E N D
Evolution of atherosclerosis VGH 1. Modification of LDL (ox-LDL) in the intima proteoglycan 2. Accumulation of LDL from oxidative stress 3. Elaboration of cytokines (interleukin-1, IL-1) 4. Recruitment of monocytes entering arterial wall (CAMs + monocyte chemoattractant protein-1, MCP-1) 5. Macrophage transformation + scanvenger receptor (M-CSF) uptake ox-LDL Foam cell 6. Migration of smooth muscle cells (SMC) 7. Extracellular matrix accumulation (MMPs) fibrosis Fatty streak 8. Programmed death (apoptosis) of SMC calcification Fibrous cap
Dysfunctional endothelial cells in Atherosclerosis VGH • Decrease in endothelium-derived relaxing factor (EDRF) promotes : • platelet aggregation, vasoconstriction, • leukocyte adhesion & increases shear • Decreasein the ratio of tissue plasminogen activator to plasminogen-activator inhibitor type 1 (t-PA / PAI-1) promotes thrombosis • Increase in adhesion molecules • promotes monocyte/macrophage retention
Extracellular matrix (ECM) VGH Smooth muscle cells (SMCs) & endothelial cells (Ecs) produce ECM Matrix metalloproteinases (MMPs) help SMCs migrate from media to intima hyperplasia Tissue inhibitors of metalloproteinases (TIMPs) delay SMC accumulation in the intima
Ischemia VGH • Supply ( low-flow) ischemia • increased vascular tone • platelet aggregation • thrombus formation • Demand ( high-flow) ischemia • chronic stable coronary obstruction • Exercise, tachycardia or emotion • flow is not meet O2 demand
The focality of lesion formation VGH Laminar shear stress augments expression of protective gene : superoxide dismutase (SOD) catabolize superoxide anion Nitric oxide synthase (NOS) produce vasodilator nitric oxide • Turbulent flow • Proximal portion of arteries after branch points • Bifurcations at flow dividers
Effects of coronary stenoses • Severity of stenosis increase flow resistance • Entrance and exit effects • Flow velocity (kinetic energy) increases • Pressure (static energy) decreases • (laminar flow eddy currents) • Lengthof stenosis • Long lesion creats eddies impact on the wall • Dynamic changes in stenosis severity • Eccentricity of atherosclerotic plaque • Vascular tone may alter luminal caliber and • stenosis resistance • stenosis > 30~45% alteration of resting flow • stenosis > 85% alteration of maximal flow VGH
Complications of Atherosclerosis Arterial stenosis VGH • Culprit lesion of Acute myocardial infarction • Antecedent coronary angiogram: • 15% of patients with stenosis > 60% • Most patients with stenosis < 50% • Coronary angiogram in post-thrombolytic cases: • 50% of patients with stenosis < 50%
Plaque Rupture VGH • 2/3 cases fracture of the fibrous cap • 1/4 cases superficial erosion of the intima • Vulnerable plaque is : • relatively lack of SMCs • foam cells and large lipid pool • macrophages in advanced athermas • overexpress matrix metalloproteins (MMPs) • & cytokines
Special cases of atherosclerosis VGH • Percutaneous transluminal coronary angioplasty • (PTCA) : • Animal proliferation of SMCs • Human constriction of adventitia • PTCA +Stenting “in-stent” srenosis • highly hydrated extracellular matrix • stellate SMCs • Graftatherosclerosis • Host T-cell interferon- SMCs growth • factor
Gross pathological changes in AMI VGH • Transmural infarcts • less sverely stenotic • more common with thrombus • single vessel • Subendocardial(non-transmural) infarcts • severely narrowed vessel • Autopsy in cases with ST segment elevation • A 90% incidence of total occlusion of culprit vessel
Microscopic features of AMI VGH • One-day coagulatiion necrosis • (nuclear shrinkage and loss, • mitochondrial damage) • wavy fibers at border • single vessel • 3rd-4th day neutrophilic infiltration • 7~10th day removal of necrotic myocytes by • macrophage • 3rd week granulation tissue with a rich • vascular network • 6th week well-healed by dense collagenous scar
Post MI ventricular remodling VGH • Residual (noninfarcted) viable myocardium • LV dilatation • cause factors loading condition • infarct artery patency • mechanism maintain stroke volume • complication nonuniform repolarization causes • life-threatening ventricular • arrhythmias • Infarct expansion • slippage between muscle bundles across • the infarct wall • disruption of normal myocytes • tissue loss within necrotic zone
VGH • Blood pressure • = peripheral resistance Xcardiac output • Cardiac output = stroke volume X heart rate • Laplace law • pressure X radius • wall tension = • wall thickness
Causes of unstable angina VGH • Plaque rupture with superimposed thrombus • 2. Dynamic obstruction • spasm of epicardial arteries • constriction of the small muscular arteries • 3. Progressive mechanical obstruction • 4. Inflammation and infection Chlamydia Pneumoniae • 5. Secondarily precipitatedby increased O2 demand • or decreased supply anemia or • thryrotoxicosis






















