Download
1 / 1
10 likes | 146 Views
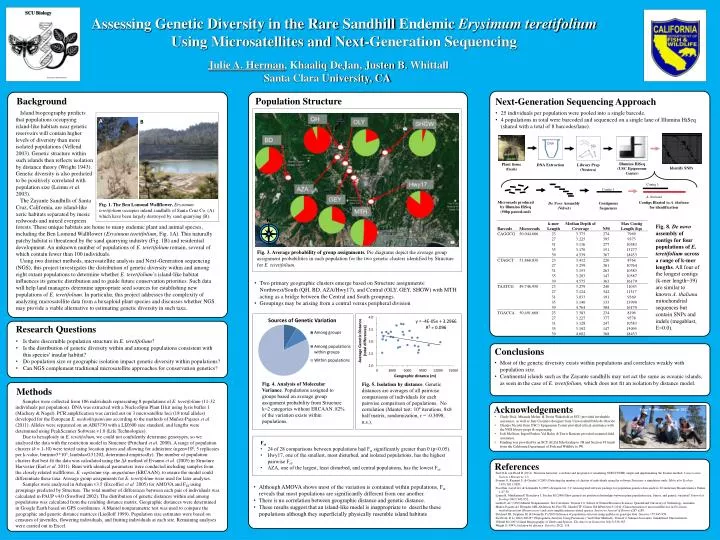
DNA. A. Microreads produced by Illumina HiSeq (50bp paired-end). Contig 1. B. A. thaliana . Contigs Blasted to A. thaliana for identification.

E N D
DNA A. Microreadsproduced by IlluminaHiSeq (50bp paired-end) Contig 1 B. A. thaliana ContigsBlasted to A. thaliana for identification Fig. 1. The Ben Lomond Wallflower. Erysimumteretifoliumoccupies inland sandhills of Santa Cruz Co. (A) which have been largely destroyed by sand quarrying (B). Assessing Genetic Diversity in the Rare Sandhill Endemic Erysimum teretifolium Using Microsatellites and Next-Generation Sequencing SCU Biology Julie A. Herman, KhaaliqDeJan, Justen B. Whittall Santa Clara University, CA Artwork by Edward Rooks Background Population Structure Next-Generation Sequencing Approach Island biogeography predicts that populations occupying island-like habitats near genetic reservoirs will contain higher levels of diversity than more isolated populations (Vellend 2003). Genetic structure within such islands then reflects isolation by distance theory (Wright 1943). Genetic diversity is also predicted to be positively correlated with population size (Leimuet al. 2003). The ZayanteSandhills of Santa Cruz, California, are island-like xeric habitats separated by mesic redwoods and mixed evergreen • 25 individuals per population were pooled into a single barcode. • 4 populations in total were barcoded and sequenced on a single lane of IlluminaHiSeq (shared with a total of 8 barcodes/lane). A B IlluminaHiSeq (USC Epigenome Center) Plant tissue (fresh) DNA Extraction Library Prep (Nextera) Identify SNPs Contig 1 De Novo Assembly (Velvet) Contiguous Sequences Fig. 8. De novo assembly of contigs for four populations of E. teretifolium across a range of k-mer lengths. All four of the longest contigs (k-mer length=39) are similar to known A. thaliana mitochondrial sequences but contain SNPs and indels (megablast, E=0.0). forests. These unique habitats are home to many endemic plant and animal species, including the Ben Lomond Wallflower (Erysimum teretifolium; Fig. 1A). This naturally patchy habitat is threatened by the sand quarrying industry (Fig. 1B) and residential development. An unknown number of populations of E. teretifolium remain, several of which contain fewer than 100 individuals. Using two distinct methods, microsatellite analysis and Next-Generation sequencing (NGS), this project investigates the distribution of genetic diversity within and among eight extant populations to determine whether E. teretifolium’s island-like habitat influences its genetic distribution and to guide future conservation priorities. Such data will help land managers determine appropriate seed sources for establishing new populations of E. teretifolium. In particular, this project addresses the complexity of analyzing microsatellite data from a hexaploid plant species and discusses whether NGS may provide a viable alternative to estimating genetic diversity in such taxa. Fig. 3. Average probability of group assignments. Pie diagrams depict the average group assignment probabilities in each population for the two genetic clusters identified by Structure for E. teretifolium. • Two primary geographic clusters emerge based on Structure assignments: Northwest/South (QH, BD, AZA/Hwy17), and Central (OLY, GEY, SHGW) with MTH acting as a bridge between the Central and South groupings. • Groupings may be arising from a central versus peripheral division Research Questions • Is there discernible population structure in E. teretifolium? • Is the distribution of genetic diversity within and among populations consistent with this species’ insular habitat? • Do population size or geographic isolation impact genetic diversity within populations? • Can NGS complement traditional microsatellite approaches for conservation genetics? Conclusions • Most of the genetic diversity exists within populations and correlates weakly with population size. • Continental islands such as the Zayantesandhills may not act the same as oceanic islands, as seen in the case of E. teretifolium, which does not fit an isolation by distance model. Acknowledgements • Cindy Dick, Miranda Melen, & Devin Wakefield at SCU provided invaluable assistance, as well as Inés Casimiro-Soriguer from Universidad Pablo de Olavide • Charles Nicolet from USC’s Epigenome Center provided critical assistance with the NGS library preps & sequencing. • Jodi McGraw, Ingrid Parker, Val Haley & TerrisKasteen provided essential field assistance. • Funding was provided by an SCU ALZA Scholarship to JH and Section VI funds from the California Department of Fish and Wildlife to JW. Fig. 4. Analysis of Molecular Variance. Populations assigned to groups based on average group assignment probability from Structure k=2 categories without ERCAAN. 82% of the variation exists within populations. Fig. 5. Isolation by distance. Genetic distances are averages of all pairwise comparisons of individuals for each pairwise comparison of populations. No correlation (Mantel test: 104 iterations, 8x8 half matrix, randomization, r = -0.3098, n.s.). Methods Samples were collected from 186 individuals representing 8 populations of E. teretifolium(11-32 individuals per population). DNA was extracted with a NucleoSpin Plant II kit using lysis buffer 1 (Machery & Nagel). PCR amplification was carried out on 3 microsatellite loci (18 total alleles) developed for the European E. mediohispanicum according to the methods of Muñoz-Pajareset al. (2011). Alleles were separated on an ABI3730 with a LIZ600 size standard, and lengths were determined using PeakScanner Software v1.0 (Life Technologies). Due to hexaploidy in E. teretifolium, we could not confidently determine genotypes, so we analyzed the data with the restriction model in Structure (Pritchard et al. 2000). A range of population clusters (k = 1-10) were tested using location priors and allowing for admixture (ngen=106, 5 replicates per k-value, burnin=5*105, lambda=0.51202, determined empirically). The number of population clusters that best fit the data was calculated using the Δk method of Evannoet al. (2005) in Structure Harvester (Earl et al. 2011). Runs with identical parameters were conducted including samples from the closely related wallflower, E. capitatum ssp. angustatum (ERCAAN), to ensure the model could differentiate these taxa. Average group assignments for E. teretifoliumwere used for later analyses. Samples were analyzed in Arlequinv3.5 (Excoffieret al. 2005) for AMOVA and FST using groupings predicted by Structure. The total number of differences between each pair of individuals was calculated in PAUP v4.0 (Swofford 2002). The distribution of genetic distances within and among populations was calculated from the resulting distance matrix. Geographic distances were determined in Google Earth based on GPS coordinates. A Mantel nonparametric test was used to compare the geographic and genetic distance matrices (Liedloff1999). Population size estimates were based on censuses of juveniles, flowering individuals, and fruiting individuals at each site. Remaining analyses were carried out in Excel. Team Wallflower, Summer 2012 References Earl D & von Holdt B (2011). Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources:1-3. Evanno G, Regnaut S, & Goudet J (2005) Detecting the number of clusters of individuals using the software Structure: a simulation study. Molecular Ecology 14(8):2611-2620. Excoffier, Laval LG, & Schneider S (2005) Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online 1:47-50. Leimu R, Mutikainen P, KorichevaJ, Fischer M (2006) How general are positive relationships between plant population size, fitness, and genetic variation? Journal of Ecology 94(5):942-952. Liedloff, AC (1999) Mantel Nonparametric Test Calculator. Version 2.0. School of Natural Resource Sciences, Queensland University of Technology, Australia. Muñoz-PajaresAJ, Herrador MB, Abdelaziz M, Picó FX, Sharbel TF, Gómez JM &Perfectti F (2011) Characterization of microsatellite loci in Erysimum mediohispanicum (Brassicaceae) and cross-amplification in related species. American Journal of Botany e287-e289. Pritchard JK, Stephens M, & Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945-959. Swofford, D L (2002) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts. VellendM (2003) Island Biogeography of Genes and Species. The American Naturalist 162(3):358-365. Wright S (1943). Isolation by distance. Genetics28(2), 114. • Fst • 24 of 28 comparisons between populations had Fst significantly greater than 0 (p<0.05). • Hwy17, one of the smallest, most disturbed, and isolated populations, has the highest pairwise Fst. • AZA, one of the largest, least disturbed, and central populations, has the lowest Fst. • Although AMOVA shows most of the variation is contained within populations, Fstreveals that most populations are significantly different from one another. • There is no correlation between geographic distance and genetic distance. • These results suggest that an island-like model is inappropriate to describe these populations although they superficially physically resemble island habitats