Download

1 / 50
500 likes | 603 Views
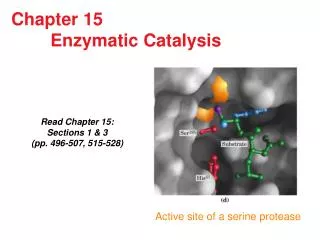
Enzyme Engineering Research & Technology Development. Enzymatic Catalysis Group, PMC Advanced Technology. Overview of objectives. Quantitative understanding of enzyme evolution (academic publications) explaining origin of natural active site sequence distributions

E N D
Enzyme Engineering Research & Technology Development Enzymatic Catalysis Group, PMC Advanced Technology
Overview of objectives Quantitative understanding of enzyme evolution (academic publications) • explaining origin of natural active site sequence distributions (benchmarking on MSA and pdb data) Redesign enzyme active sites (designer enzyme products) • modify substrate selectivity, product inhibition, etc • for industrial biocatalysis, biotechnology and biotherapeutics (with experiment) To advance the state-of-the-art in enzyme designtechnology (design software) • through the application of high-resolution physics-based methods for active site modeling using: 1) High-res protein structure prediction (OPLS + SGB): loop prediction for reshaping active sites, side chain optimization 2)Semiempirical enzyme-substrate binding affinity scoring (Km), substrate pose sampling 3) Refinement based on details of electronic structure: scoring activation energies (kcat)
Schematic of computational enzyme design technology Software Patents Design Protocol Patents
Design Computationally Refine Experimentally Input information Target chemical Desired raw material Existing synthetic pathways Existing biocatalysts System Output ~1000 potential candidates expected catalytic activity Optimized Biocatalyst Zymzyne™ Computational Design Process Zymzyne™ Experimental Optimization 1030 candidates screened 500 candidates screened Zymzyne Enzyme Design and Optimization Platform Design Protocol Patents Software Patents
A model fitness measure for enzyme sequence optimization substrate binding catalysis product release • Maximize free energy of substrate binding over sequence space • Represent catalysis through constraints on interatomic • distances of catalytic side chains • Minimize total energy of complex for any sequence • To start, omit selection pressure for product release
Active site sequence optimization requires accurate energy functions, solvation models, and search algorithms 10o resolution rotamer library (297 proteins) Xiang, Z. and Honig, B. (2001) J. Mol. Biol.311: 421-430.
Active site sequence optimization requires accurate energy functions, solvation models, and search algorithms S-GB continuum solvation 10o resolution rotamer library (297 proteins) Ghosh, A., Rapp, C.S. & Friesner, R.A. (1998) J. Phys Chem. B102, 10983-10990. Xiang, Z. and Honig, B. (2001) J. Mol. Biol.311: 421-430.
Active site sequence optimization requires accurate energy functions, solvation models, and search algorithms S-GB continuum solvation 10o resolution rotamer library (297 proteins) Ghosh, A., Rapp, C.S. & Friesner, R.A. (1998) J. Phys Chem. B102, 10983-10990. Xiang, Z. and Honig, B. (2001) J. Mol. Biol.311: 421-430. OPLS-AA molecular mechanics force field + Glidescore semiempirical binding affinity scoring function Friesner, R.A, Banks, J.L., Murphy, R.B., Halgren, T.A. et al. (2004) J. Med. Chem. 47, 1739-1749. Jacobson, M.P., Kaminski, G.A. Rapp, C.S. & Friesner, R.A. (2002) J. Phys. Chem. B106, 11673-11680.
φ,ψ = the backbone torsion angles Backbone = the sequence of (COOH)-[N-(CH-Ri)-(C=O)]N-NH2 , where Ri is the i'th side chain. 2N torsion angles specify the backbone configuration. Side-chains have their own rotamers too! These angles are represented by χi. Some side chains have no χ angles. Some have quite a few, such as the lysine above with χ1-χ4.
Computational sequence optimization correctly predicts most residues in ligand-binding sites… StreptavidinNative –10.04 kcal/mol Chakrabarti, R., Klibanov, A.M. and Friesner, R.A. Computational prediction of native protein ligand-binding and enzyme active site sequences. PNAS, 2005.
Computational sequence optimization correctly predicts most residues in ligand-binding sites… StreptavidinNative –10.04 kcal/mol Chakrabarti, R., Klibanov, A.M. and Friesner, R.A. Computational prediction of native protein ligand-binding and enzyme active site sequences. PNAS, 2005.
Computational sequence optimization correctly predicts most residues in ligand-binding sites… StreptavidinNative –10.04 kcal/mol CO2- is covalent attachment site for biomolecules 9 / 10 residues predicted correctly in top 0.5 kcal/mol of sequences Easy to exptly screen libraries of this size Chakrabarti, R., Klibanov, A.M. and Friesner, R.A. Computational prediction of native protein ligand-binding and enzyme active site sequences. PNAS, 2005.
…and enzyme active sites R61 DD-peptidaseNative –10.02 kcal/mol
…and enzyme active sites R61 DD-peptidaseNative –10.02 kcal/mol High MSA variability T123 highly degenerate in multiple sequence alignment
Computational enzyme sequence optimization: sugar catalysis b-galactosidaseNative –9.13 kcal/mol Computed • Native amino acid is generally one of • top 3 most frequently predicted • Could be used to focus combinatorial libraries • (3N vs 20N, N = # of residues)
Computed amino acid distributions contain detailed evolutionary information Glucose-binding proteinNative –8.81 kcal/mol
Computed amino acid distributions contain detailed evolutionary information Observed (sequence alignment) Glucose-binding proteinNative –8.81 kcal/mol
Computed amino acid distributions contain detailed evolutionary information Observed (sequence alignment) Glucose-binding proteinNative –8.81 kcal/mol Epimeric promiscuity Anomeric promiscuity Computed OH OH • Computed residue frequencies often mirror • natural frequencies
Summary of recent results: classical sequence optimization (Side chain prediction/ Binding affinity calculation / Sequence opt) Acid/base Y159 Electrostatic stabilizer Lys65 Nucleophile Ser62 T123 highly degenerate in multiple sequence alignment R61 DD-peptidase
Computed amino acid distributions contain detailed evolutionary information Observed (sequence alignment) Glucose-binding proteinNative –8.81 kcal/mol Epimeric promiscuity Anomeric promiscuity Computed OH OH • Computed residue frequencies often mirror • natural frequencies
High-resolution sequence optimization is robust across diverse functional families Peptide Nucleotide Sugar
Active Site Design of Enzymes with Nucleotide Substrates: Cytidine Kinase Cytidine kinase native pose
Multisubstrate enzyme active site sequences represent superpositions of computational predictions dTMP HSV-1 thymidine kinase
Multisubstrate enzyme active site sequences represent superpositions of computational predictions dTMP Ganciclovir (dG analog)
Multisubstrate enzyme active site sequences represent superpositions of computational predictions dTMP Apply multiobjective sequence search algorithms to accommodate several substrates Ganciclovir (dG analog) Thymidine
Multisubstrate enzyme active site sequences represent superpositions of computational predictions dTMP Ganciclovir (dG analog) Native sequence = superposition of optimal sequences for multiple substrates Thymidine
Catalytic hydrogen-bonding networks can be incorporated into sequence optimization GLU 272 LYS 315 Cephalothin W402 d b c SER 62 b-Lactamase : cephalothin e g GLU 272 a f h LYS 67 TYR 150 ARG 148 GLN 120 ASN 152
Catalytic hydrogen-bonding networks can be incorporated into sequence optimization +2 kcal/mol +1 kcal/mol Constrained Constrained + Filtered LYS 315 Cephalothin W402 d b c SER 62 e g GLU 272 a f h LYS 67 TYR 150 ARG 148 GLN 120 ASN 152 Chakrabarti, R., Klibanov, A.M. and Friesner, R.A. Sequence optimization and designability of enzyme active sites. PNAS, 2005.
Refining the scoring function: quantum chemical transition state calculations Enzyme kcat (s-1) KM (μM) kcat/KM (% Wild-type) WT 150 14 100 N152S 3 7 4.3 N152D 0.12 24 0.05 N152S/Q120F 3 4.6 6.7 N152S/Q120H 20 11.4 16.3 Predicted 14.3 kcal/mol Measured 14.3 kcal/mol
Active Site Designability: The Number of Sequences that Solve a Given Design Problem +2 kcal/mol + 1 kcal/mol Constrained Number of residues correctly predicted General acid/base Y159 Electrostatic stabilizer Lys65 Catalytic nucleophile Glu-299 Catalytic Nucleophile Ser62 General acid/base Glu-200 DD-peptidase b-gal
Patents: computational sequence optimization / experimental mutagenesis Example of screening focused library of sequence variants 3 permissible mutations identified by modeling at a target position 3 positions subject to mutagenesis 43 mutation combinations = 64 sequence variations Synthetic gene assembly and variant library construction via DNA synthesis Biological selection of variant library New enzymes - Improved catalytic turnover Altered substrate selectivity Ask for details from Zhen and/or NEB; limitations on how libraries are made, how many sequences can be screened? Chakrabarti, R., De Jong, R., Cornish, V.C. and Friesner, R.A., unpublished results
Patents: algorithms in development Protein structure Substrate binding Reactive chemistry Possibly show any visios – ask group members to convert. Plan next steps Loop Sidechain Glidescore Pose sampling New algorithms for side chain optimization QM sequence refinement Classical Sequence Optimization (fixed ligand) Red = internal to code Black = script • for QM/MM refinement • of enzyme design • speeding up mutant • TS searches Calculating mutant enzyme reaction rates Classical Sequence Optimization (free ligand) Active site reshaping • Hierarchical pose screening • Locates global seq/struct optima • for a given active site/ligand comb • Estimates “designability” of active site • (fixed backbone) • scores desired loop • against other low-energy • excitations
Testing: Current experimental projects • Dehalogenase-dehydrogenase redesign: arbitrary backbone reshaping to • accommodate NAD – being tested now, could also benefit from faster side chain opt • Art testing induced fit results on this system without sequence opt • PBP/ b-lactamase redesign (needed – covalent docking in Glide [XP grow] or Macromodel-prime): • helix breaking (now being done)/backbone reshaping + redocking + QM/MM sequence refinement • Single mutation activation barrier predictions in b-lactamase – currently being tested
Discussion Points • NEB combinatorial screening protocols • NEB DNA enzyme engineering challenge problems • Scope for interaction: • Technology Platform to be used by both parties? • Engineered DNA Enzyme Products? Cosolvent-resistant polymerases? • IP: Software, Designability-Based Screening Protocols (compare Maxygen, Diversa), and Engineered Enzymes
Sirtuin – mutant production, selection, protein expression and enzyme assay • Main objective - Develop genetic and biochemical assay systems to screen sirtuin mutant library and quantify enzymatic activities. • Main steps - • Model mutations in the active site residues of bacterial sir2Tm. • Generate a set of mutations using wild-type sirtuin as template based on computation-guided structural modeling. • Transform the mutants into host strains with sirtuin deletion. • Assay growth of mutant transformants under carbon source limitation. • Select mutant constructs which can complement the growth defects resulted from sirtuin deficiency, which are manifested under carbon limitation. • Purify the wild-type and active mutant enzymes and quantify their kinetic properties.
Sirtuin – mutant generation Model mutations in the active sites of sirtuin genes by computational analysis. Construct mutations in the wild-type sir2Tm plasmid (2 potential methods) By synthetic gene method – Generate sequence map for proposed nucleotide changes in the wild-type template (sir2Tm) . Work with gene synthesis groups to make synthetic constructs for the mutant collection, e.g. how to get efficient oligo assembly to cover all the mutations. Obtain suitable plasmid vectors and clone the mutant constructs into the vectors. The vectors would depend on the host cells in which the mutant constructs would be expressed and selected, e.g. yeast, salmonella have different vectors to allow high-level expression. By multi-site directed mutagenesis method – Use reagents including cells, enzymes and mutagenic primers to generate mutation in the wild-type sirTm template. Verify mutations by DNA sequencing. Both procedures for mutagenesis depend on the actual mutations to be made and how many constructs are needed to allow for effective functional screening.
Sirtuin – mutant library screening assay When the mutant collection is generated, transform the constructs into host cell with sirtuin deletion. Make competent cells for the host strain so they can take up DNA. Transform the wild-type plasmid into host as positive control. Transform the mutant plasmids into host cells. Assay whether the transformants could grow on carbon-limited media, such as with acetate or propionate as sources. If there is complementation, characterize the growth features of these cells. Verify the specific mutations by DNA sequencing. Transform the mutant construct into protein-expression host, such as Ecoli BL21. Grow cultures and purify sufficient quantities of proteins. Set up enzymatic assays to quantify kinetic properties of wild-type and selected mutants.
Beta-lactamase – mutant selection, protein expression and activity assay • Model mutations in the active site residues of P99 beta-lactamase. • Construct mutations in the wild-type P99 beta-lactamase gene. • Obtain bacterial host strains suitable for screening beta-lactam antibiotic resistance. • Transform bacteria host cells with wild-type and mutant constructs. • Select transformed cells in the presence of beta-lactam antibiotics. • Identify the mutant clones which can grow in beta-lactam and thus retain beta-lactamase activities. • Express and purify the wild-type and mutant beta-lactamases and quantify their kinetic properties.
Beta-lactamase – mutant generation Model mutations in the active site of P99 beta-lactamase based on computation. Construct mutations in the wild-type P99 beta-lactamase plasmid. The actual processes would depend on what the mutations are and how many mutants are to be made. By synthetic gene method – Work with gene synthesis group to construct synthetic constructs, esp. in how to set up efficient oligonucleotides coverage for all the mutations. Clone all mutant constructs into suitable bacterial expression vector. By multi-site directed mutagenesis method - Need to obtain mutagenic reagents such as cells, enzymes and primers to generate a set of mutations. Verify mutant production by DNA sequencing of individual clones.
Beta-lactamase – mutant selection With the bacterial host strains used for selection, make competent cells so that they can take up plasmid DNA. Transform wild-type P99 beta-lactamase plasmid into host cells as positive control. Transform the mutant plasmids into host cells to select for active constructs. Make agar plates containing different types of beta-lactam compounds and at different concentration. Grow bacteria transformed with beta-lactamase plasmids on these plates and monitor colony formation. Identify the clones with good growth characteristics so they would be the candidates to provide hydrolytic activities on a variety of beta-lactam substrates. Verify specific mutations by DNA sequencing. Proceed to protein expression, purification and activity quantitation.
A General Framework for Computationally Directed Biocatalyst Design slack variable [may list other fitness measures] Catalytic constraint: interatomic distances rij < hbond dist Enzyme-substrate binding affinity • Minimize J over sequence space • Represent dynamical constraint with requirement that total energy of complex • minimized for any sequence • Omits selection pressure for product release
Assessment of active site designability • Need to assess number of sequences that are structurally similar to native • Requires sampling over ligand conformations
Computationally directed active site sequence library generation • Two approaches: • Marginal distributions (as shown) using top m (m constant) as shown or setting m_i according to exp(shannon entropy). Choose T based on exptl tractability. Assumes independence, but easier for exptlst to implement out-of-box. Note S in this case cannot be interpreted as number of microstates since LLN does not hold • b) Joint distribution: sample m sequences from joint distribution for specified T’s. S computed based on moments of objectives. Compare D=exp(S) for several T’s, look for transition to region where denser sampling possible (heat capacity analogy). LLN holds, allowing interpretation of designability as relative number of microstates
Computed sequence entropies suggest equilibrium in sequence space Comparable Shannon site entropies suggest equilibrium for same fitness measure and provide concise comparison of distributions at all positions (rather than showing pdf at each position) Shannon sequence entropy: Si = - S(a=1...20) [f(ia) ln f(ia)] Computed Observed Catalytic constraint Penicillium sp. b-galactosidase
Marginal active site sequence distributions Shannon site entropies: Computed based on marginal distributions; unlike joint cannot be expressed in closed form in terms of exp fn. Two approaches to estimating distribution – a) in terms of marginal moments of functions of f_i’s; b) in terms of explicit f_i’s (used here). Both based on drawing m samples from joint Extensions/modifications to PNAS paper figures: Better to display K-L relative entropies rather than site entropies for marginal distributions at each position Instead compare K-L relative entropies (joint distribution) wrt MSAs for models w different objectives, on same plot; alternatively use approach based on marginal distributions on Shannon entropy slide For such figures, compare K-L rel entropies (here marginal)
Plan for development of designability theory and experimental application (to be described in conclusion of our early papers) • Apply designability theory to all major enzyme families from PNAS papers; extend to designability of modified sirtuins experimentally • Could id the catalytic constraints and focus on objective for reducing inhibition (NAM binding affinity); estimate latter temperature. • Compare designability of NAD site to that of other enzyme classes studied, for same T’s. Check designability at lower T for NAM inhibition • Designability approach will help determine viability of drug development efforts more effectively than comb chem
Components of energy function Surface-area term Covalent bond potential Torsional potential H2O H2O H-bonding (sometimes) H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O H2O Non-bonding terms (Van Der Waals) The effect of water (a rude fellow!) Electrostatic potential http://tinyurl.com/63gt3lm
Computational active site optimization is structurally accurate to near-crystallographic resolution
Future plans • Understanding differences between PLOP/Glide/Qsite energies for summing energy calcs to calculate Km, kcat • Modeling the denatured state of proteins to estimate folding free energy for core sequence optimization Integration with other current developments • Induced fit + Backbone reshaping to start with globally-relaxed backbone shapes for unnatural ligands • MD treatment of loops + Backbone reshaping + Classical affinity opt for antibody engineering