Download

1 / 7
70 likes | 86 Views
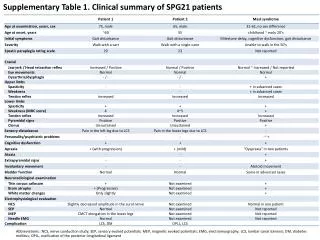
This supplementary table provides a clinical summary and cognitive function assessment of patients with SPG21, including nerve conduction studies, sensory evoked potentials, and more. It also includes data on age at onset and mutations in the SPAST gene.

E N D
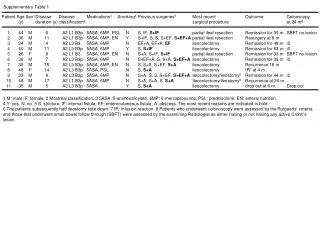
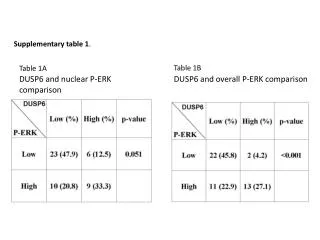
Supplementary Table 1. Clinical summary of SPG21 patients Abbreviations:: NCS, nerve conduction study; SEP, sensory evoked potentials; MEP, magnetic evoked potentials; EMG, electromyography; LCS, lumbar canal stenosis; DM, diabetes mellitus; OPLL, ossification of the posterior longitudinal ligament
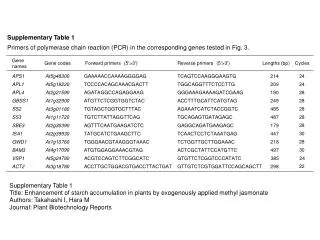
Supplementary Table 2. Cognitive function of SPG21 patients Abbreviations: MMSE, Mini-mental state examination FAB, frontal assessment battery WAIS-R, Wechsler adult intelligence scale-revised IQ, intelligence quotient VIQ, verbal IQ PIQ, performance IQ
Supplementary Fig. 1:Distribution of age at onset A Distribution of age at onset of patients classified on the basis of clinical phenotype n=127 Number of patients Age at onset B Distribution of age at onset of patients classified on the basis of family history n=127 Number of patients Age at onset
Supplementary Figure 1. Distribution of age at onset according to phenotype and family history (A) The distribution of age at onset of 127 index patients whose detailed clinical data are obtained is classified on the basis of clinical phenotype. In pure-form HSP patients, bimodal peaks of the distribution in the first and the 5th decades are noted, whereas a large peak in the first decade and a small peak in the 50s and 60s were found in the complicated-form HSP patients. (B)The distribution of age at onset of 127 patients on the basis of family history. The ages at onset of both AD-HSP patients and sporadic HSP patients show similar bimodal distributions, whereas AR-HSP patients tend to show an early onset. (C) Distribution of the age at onset in SPG4 patients (n=38) according to the type of mutation. In addition to index patients, we include affected family members when precise clinical information is available. The age at onset does not seem to correlate with the type of mutation in SPAST.
Supplementary Fig. 2: Mutations in SPASTidentified in the present study A Del ex17* Rearrangements Dup ex5-7* Del ex17* Del ex1* Del ex2-17* * Nonsense mutations * * Null mutations * * * * * * * * * * * Frameshift mutations Exon 1 Exon 17 Splicing mutations Missense mutations 1 342 599 616 AAA casette * Novel mutations * Missense mutations * * B Y52C T369P L549P H. sapiens PPPESPH-KRNLYYFSYPLF PELFTGLRA SGSDLTALA P. troglodytes PPPESPH-KRNLYYFSYPLF PELFTGLRA SGSDLTALA R. norvegicus PAPGSPH-KRNLYYFSYPLV PELFTGLRA SGSDLTALA M. musculus PAAGSPP-KRNPSSFSSPLV PELFTGLRA SGSDLTALA G. gallus AAAASPH-KRNLYYFSYPLF PELFTGLRA SGSDLTALV X. tropicalis LAPPSLH-KRNLYLFSYPLL PELFTGLRA SGSDITALA D. rerio ----SARGNRLLFYTRSLSR PELFTGLRA SGSDLTSLA D. melanogaster ----SVH-KQNLYVVSFPII PELFTGLRA SGSDLTALA #E43Q #P45Q previously described missense mutations in exon 1 #P41L #S44L
Supplementary Figure 2. Mutations in SPAST identified in the present study (A) Spastinis a 616-amino-acid protein, transcribed from 17 exons. Positions of nonsense (green squares), frameshift (blue circles), splicing (purple diamonds), missense (light blue triangles), and rearrangement (orange lines) mutations detected in this study are indicated. Nineteen novel mutations detected in the study were indicated by a star. Nonsense, frameshift, or splice site mutations (24/32 families) are distributed throughout the genes, whereas most missense mutations in SPAST (7/8 families) are located in ATPase associated with various cellular activities (AAA) domain. (B)Analysis of evolutionally conserved sequences in spastin and comparison with novel mutations (Y52C, T369P, and L549P) detected in this study (underlined). Two mutations in the AAA domain (T369P and L549P) affect highly conserved amino acids. Y52 is outside the AAA domain, located near four previously reported amino acid substitutions in exon 1 of SPAST (indicated by sharps). Y52C involves an evolutionally conserved amino acid among chimpanzees, rats, chickens, and zebrafish, and is located in a putative nuclear export signal. The patient with the heterozygous Y52C substitution, born to consanguineous parents, suffered from cervical canal stenosis, and the symptoms gradually progressed even after a neck surgery. One of the healthy parents carried the heterozygous Y52C substitution. Y52C was neither present in the controls nor reported in the dbSNP database (http://www.ncbi.nlm.nih.gov/snp), raising a possibility that it is a mutation with reduced penetrance. However, we cannot completely exclude the possibility that Y52C is a rare variant with little relevance to the patient’s neurological conditions. Abbreviations: Dup, duplication; Del, deletion
Supplementary Fig. 3: Distribution of age at onset of HSP in patients with SPG4 and SPG11 A Patients with mutations in SPAST (SPG4) n=38 Number of patients Age at onset B Patients with mutations in SPG11 (SPG11) Number of patients Age at onset (A) Age at onset of HSP in patients with SPG4 showed bimodal distribution. In addition to data of all the index patients, those of the family members whose clinical information was available were included (n=38). Types of mutations did not correlate with age at onset. (B) The ages at onset of HSP in patients with SPG11 were under 30 years old.