Download

1 / 77
800 likes | 1.05k Views
High-Throughput Sequencing. Richard Mott with contributions from Gil Mcvean , Gerton Lunter , Zam Iqbal , Xiangchao Gan , Eric Belfield. Sequencing Technologies. Capillary ( eg ABI 3700) Roche/454 FLX Illumina GAII ABI Solid Others…. Capillary Sequencing. based on electrophoresis

E N D
High-Throughput Sequencing Richard Mott with contributions from Gil Mcvean, GertonLunter, ZamIqbal, XiangchaoGan, Eric Belfield
Sequencing Technologies • Capillary (eg ABI 3700) • Roche/454 FLX • Illumina GAII • ABI Solid • Others….
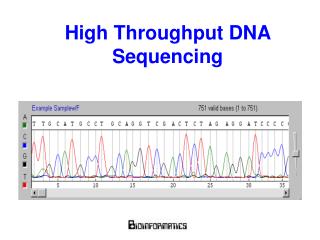
Capillary Sequencing • based on electrophoresis • used to sequence human and mouse genomes • read lengths currently around 600bp (but used to be 200-400 bp) • relatively slow – 384 sequences per run in x hours • expensive ???
Capillary Sequencing Trace http://wheat.pw.usda.gov/genome/sequence/ ACGT represented by continuous traces. Base-calling requires the identification of well-defined peaks
PHRED Quality Scores • PHRED is an accurate base-caller used for capillary traces (Ewing et al Genome Research 1998) • Each called base is given a quality score Q • Quality based on simple metrics (such as peak spacing) callibrated against a database of hand-edited data • Q = 10 * log10(estimated probability call is wrong) 10 prob = 0.1 20 prob = 0.01 30 prob = 0.001 [Q30 often used as a threshold for useful sequence data]
Capillary sequence assembly and editing CONSED screen shots http://www.jgi.doe.gov/education/how/how_12.html
Illumina Sequencing machines GA-II HiSeq
The Illumina Flow-cell • Each flow-cell has 8 lanes (16 on HiSeq) • A different sample can be run in each lane • It it possible to multiplex up to 12 samples in a lane • Each lane comprises 2*60=120 • square tiles • Each tile is imaged and analysed separately • Sometimes a control phiX lane is run (in a control, the genome sequenced is identical to the reference and its GC content is not too far from 50%)
Illumina GA-II “traces” Discontinuous – a set of 4 intensities at each base position
Cross talk: base-calling errors Whiteford et al Bioinformatics 2009
Base-calling errors Typical base-calling error rate ~ 1%, Error rate increases towards end of read Usually read2 has more errors than read1
Assessing Sequence Quality Example summary for a lane of 51bp paired-end data # reads % optical duplicates % good quality reads (passed chastity filter) % mapped to reference using ELAND (the read-mapper supplied by Illumina) % of differences from reference (upper bound on error rate) in mapped reads
Illumina Throughput (April 2013) Note: 1 human genome = 3Gb. 20-30x coverage of one human genome = 2=3 lanes of HiSeq
Illumina HiSeqThroughput (Feb 2011) Note: 1 human genome = 3Gb.
Illumina GA-II throughput per flow-cell Note: these are correct as of February 2010. Output is constantly improving due to changes in chemistry and software. Consumables costs are indicative only - they don’t include labour, depreciation, overheads or bioinformatics - true costs are roughly double
Pooling and Multiplexing Primer Primer Read Barcode 6bp Up to 96 distinct barcodes can be added to one end of a read useful for low-coverage sequencing of many samples in a simple lane Up to a further 96 barcodes can be added to other end of a read = 96*96 = 9216 samples Useful for bacterial sequencing
Pooling Costs • Library Preps • £80-150 per sample, depending on type of sequencing • £<50 per sample for 96-plex genomic DNA • Pooling costs are dominated by library prep, not HiSeq lane costs • eg 96-plex of gDNA on on HiSeq lane = £4k
Data Formats • Sequencing produces vast amounts of data • Rate of data growth exceeds Moore’s law
The FastQ format(standard text representation of short reads) • A FASTQ text file normally uses four lines per sequence. • Line 1 begins with a '@' character and is followed by a sequence identifier and an optional description (like a FASTA title line). • Line 2 is the raw sequence letters. • Line 3 begins with a '+' character and is optionally followed by the same sequence identifier (and any description) again. • Line 4 encodes the quality values for the sequence in Line 2, and must contain the same number of symbols as letters in the sequence. The letters encode Phred Quality Scores from 0 to 93 using ASCII 33 to 126 • Example • @SEQ_ID • GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT • + • !''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
Binary FastQ • Computer-readable compressed form of FASTQ • About 1/3 size of FASTQ • Enables much faster reading and writing • Standard utility programs will interconvert (eg. maq) • Becoming obsolete……
SAM and SAMTOOLShttp://samtools.sourceforge.net/ • SAM (Sequence Alignment/Map) format is a generic format for storing large nucleotide sequence alignments. • SAM aims to be a format that: • Is flexible enough to store all the alignment information generated by various alignment programs; • Is simple enough to be easily generated by alignment programs or converted from existing alignment formats; • Is compact in file size; • Allows most of operations on the alignment to work on a stream without loading the whole alignment into memory; • Allows the file to be indexed by genomic position to efficiently retrieve all reads aligning to a locus. • SAM Tools provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format.
BAM files • SAM, BAM are equivalent formats for describing alignments of reads to a reference genome • SAM: text • BAM: compressed binary, indexed, so it is possible to access reads mapping to a segment without decompressing the entire file • BAM is used by lookseq, IGV and other software • Current Standard Binary Format • Contains: • Meta Information (read groups, algorithm details) • Sequence and Quality Scores • Alignment information • one alignment per read
Inside a BAM file @HD VN:1.0 GO:noneSO:coordinate @SQ SN:chr10 LN:135534747 @SQ SN:chr11 LN:135006516 ... @SQ SN:chrX LN:155270560 @SQ SN:chrY LN:59373566 @RG ID:WTCHG_7618 PL:ILLUMINA PU:101001_GAII06_00018_FC_5 LB:070/10_MPX SM:1772/10 CN:WTCHG @PG ID:stampy VN:1.0.5_(r710) CL:--processpart=1/4 --readgroup=ID:WTCHG_7618,SM:1772/10,PL:ILLUMINA,PU:101001_GAII06_00018_FC_5,LB:070/10_MPX,CN:WTCHG --comment=@Lane_5_comments.txt --keepreforder --solexa -v0 -g /tmp/Human37 -h /tmp/Human37 -M s_5_1_sequence.txt,s_5_2_sequence.txt --bwaoptions=-t 2 -q10 /tmp/Human37 -o s_5.1_stampy.sam @PG ID:stampy.1 VN:1.0.5_(r710) CL:--processpart=2/4 --readgroup=ID:WTCHG_7618,SM:1772/10,PL:ILLUMINA,PU:101001_GAII06_00018_FC_5,LB:070/10_MPX,CN:WTCHG --comment=@Lane_5_comments.txt --keepreforder --solexa -v0 -g /tmp/Human37 -h /tmp/Human37 -M s_5_1_sequence.txt,s_5_2_sequence.txt --bwaoptions=-t 2 -q10 /tmp/Human37 -o s_5.2_stampy.sam @PG ID:stampy.2 VN:1.0.5_(r710) CL:--processpart=3/4 --readgroup=ID:WTCHG_7618,SM:1772/10,PL:ILLUMINA,PU:101001_GAII06_00018_FC_5,LB:070/10_MPX,CN:WTCHG --comment=@Lane_5_comments.txt --keepreforder --solexa -v0 -g /tmp/Human37 -h /tmp/Human37 -M s_5_1_sequence.txt,s_5_2_sequence.txt --bwaoptions=-t 2 -q10 /tmp/Human37 -o s_5.3_stampy.sam @PG ID:stampy.3 VN:1.0.5_(r710) CL:--processpart=4/4 --readgroup=ID:WTCHG_7618,SM:1772/10,PL:ILLUMINA,PU:101001_GAII06_00018_FC_5,LB:070/10_MPX,CN:WTCHG --comment=@Lane_5_comments.txt --keepreforder --solexa -v0 -g /tmp/Human37 -h /tmp/Human37 -M s_5_1_sequence.txt,s_5_2_sequence.txt --bwaoptions=-t 2 -q10 /tmp/Human37 -o s_5.4_stampy.sam @CO TM:Tue, 26 Oct 2010 09:21:06 BST WD:/data1/GA-DATA/101001_GAII06_00018_FC/Data/Intensities/BaseCalls/Demultiplexed-101009/004/GERALD_09-10-2010_johnb.2 HN:comp04.well.ox.ac.uk UN:johnb @CO IX:GCCAAT SN:085 B-cell ID:070/10_MPX GE:Human37 SR:gDNA Indexed CT:false PR:P100116 SM:1771/10 @CO TM:Tue, 26 Oct 2010 09:21:06 BST WD:/data1/GA-DATA/101001_GAII06_00018_FC/Data/Intensities/BaseCalls/Demultiplexed-101009/004/GERALD_09-10-2010_johnb.2 HN:comp04.well.ox.ac.uk UN:johnb @CO IX:GCCAAT SN:085 B-cell ID:070/10_MPX GE:Human37 SR:gDNA Indexed CT:false PR:P100116 SM:1771/10 @CO TM:Tue, 26 Oct 2010 09:21:06 BST WD:/data1/GA-DATA/101001_GAII06_00018_FC/Data/Intensities/BaseCalls/Demultiplexed-101009/004/GERALD_09-10-2010_johnb.2 HN:comp04.well.ox.ac.uk UN:johnb @CO IX:GCCAAT SN:085 B-cell ID:070/10_MPX GE:Human37 SR:gDNA Indexed CT:false PR:P100116 SM:1771/10 @CO TM:Tue, 26 Oct 2010 09:21:06 BST WD:/data1/GA-DATA/101001_GAII06_00018_FC/Data/Intensities/BaseCalls/Demultiplexed-101009/004/GERALD_09-10-2010_johnb.2 HN:comp04.well.ox.ac.uk UN:johnb @CO IX:GCCAAT SN:085 B-cell ID:070/10_MPX GE:Human37 SR:gDNA Indexed CT:false PR:P100116 SM:1771/10 WTCHG_7618:5:40:5848:3669#GCCAAT 145 chr10 69795 3 11I40M chr16 24580964 0 TCAGAAAAAAGAAAATGTGGTATATATACACAATGGAGTACTATTCAGCCC GFIIIIIIIIHIIIIHIIIIIIIIFIIHIIHIIHIIIIIHIIIIIIIHHII PQ:i:394 SM:i:0 UQ:i:250 MQ:i:96 XQ:i:40 RG:Z:WTCHG_7618 WTCHG_7618:5:77:5375:15942#GCCAAT 99 chr10 82805 0 51M = 83055 301 GCAGGGAGAATGGAACCAAGTTGGAAAACACTCTGCAGGATATTATCCAGG GBBHHEBG<GGGGGGEGGGEGDGDGGDBGDHHHFHGGEGEBGGDGHEHHFH PQ:i:91 SM:i:0 UQ:i:38 MQ:i:0 XQ:i:33 RG:Z:WTCHG_7618 WTCHG_7618:5:77:5375:15942#GCCAAT 147 chr10 83055 0 51M = 82805 -301 AGCTGATCTCTCAGCAGAAACCGTACAAGCCAGAAGAGAGTGGGGGCCAAC #################DB-B?B8B>G>GGBID?FHBBI@IGGGGEEDGGG PQ:i:91 SM:i:0 UQ:i:33 MQ:i:0 XQ:i:38 RG:Z:WTCHG_7618 WTCHG_7618:5:49:18524:13016#GCCAAT 163 chr10 83516 0 51M = 83734 269 CCCATCTCACGTGCAGAGACACACATAGACTCAAAATAAAAGGATGGAGGA EHHIIIHIIIIHIIIIIFIDIIIEGEIIIHIHIIIIIIHHIHBDHEGFDEI PQ:i:57 SM:i:0 UQ:i:0 MQ:i:0 XQ:i:39 RG:Z:WTCHG_7618 WTCHG_7618:5:2:1789:11020#GCCAAT 161 chr10 83598 0 2M2D49M chrM 2220 0 GTGGGTTGCAATCCTAGTCTCTGATAAAACAGACTTTAAACCAATAAAGAT GGGGG>DDBGGGGGGIIGIBDFGE?IIDIHIIIIBIGIBIHIIHII<DAI< PQ:i:192 SM:i:0 UQ:i:48 MQ:i:96 XQ:i:0 RG:Z:WTCHG_7618 WTCHG_7618:5:5:8834:6028#GCCAAT 163 chr10 83702 0 51M = 83876 225 AGAAGAGCTAACTATCCTAAATATATATGCACCCAATACAGGAGCACCCAG EIIIIHHHGGIDIIIHEGIGIHGIGIDFIGBGGGEGGGGGIHDHIDIIHGH PQ:i:23 SM:i:0 UQ:i:0 MQ:i:0 XQ:i:0 RG:Z:WTCHG_7618 WTCHG_7618:5:49:18524:13016#GCCAAT 83 chr10 83734 0 51M = 83516 -269 CCAATACAGGAGCACCCAGATTCATAAAGCAAGTCCTGAGTGACCTACAAT BHHHHGHHHHHHHHFHHHHHHHGHHHGGGGBHHEHHHHHHHHHHHHHHHHH PQ:i:57 SM:i:0 UQ:i:39 MQ:i:0 XQ:i:0 RG:Z:WTCHG_7618 WTCHG_7618:5:5:8834:6028#GCCAAT 83 chr10 83876 0 51M = 83702 -225 TACCCAGGAATTGAACTCAGCTCTGCACCAAGCAGACCTAATAGACATCTA DEHIIIHIIIIDIGIHFHHGIHIGIIIIIIIIHIGIIIHIIHIIIIIIGII PQ:i:23 SM:i:0 UQ:i:0 MQ:i:0 XQ:i:0 RG:Z:WTCHG_7618 samtools view -h WTCHG_7618.bam
SAMtools • A package for manipulating sequence data • import: SAM-to-BAM conversion • view: BAM-to-SAM conversion and subalignment retrieval • sort: sorting alignment • merge: merging multiple sorted alignments • index: indexing sorted alignment • faidx: FASTA indexing and subsequence retrieval • tview: text alignment viewer • pileup: generating position-based output and consensus/indel calling • Li H.*, Handsaker B.*, Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 25, 2078-9
Pileup Alignments seq1 272 T 24 ,.$.....,,.,.,...,,,.,..^+. <<<+;<<<<<<<<<<<=<;<;7<& seq1 273 T 23 ,.....,,.,.,...,,,.,..A <<<;<<<<<<<<<3<=<<<;<<+ seq1 274 T 23 ,.$....,,.,.,...,,,.,... 7<7;<;<<<<<<<<<=<;<;<<6 seq1 275 A 23 ,$....,,.,.,...,,,.,...^l. <+;9*<<<<<<<<<=<<:;<<<< seq1 276 G 22 ...T,,.,.,...,,,.,.... 33;+<<7=7<<7<&<<1;<<6< seq1 277 T 22 ....,,.,.,.C.,,,.,..G. +7<;<<<<<<<&<=<<:;<<&< seq1 278 G 23 ....,,.,.,...,,,.,....^k. %38*<<;<7<<7<=<<<;<<<<< seq1 279 C 23 A..T,,.,.,...,,,.,..... ;75&<<<<<<<<<=<<<9<<:<<
Genome Resequencing • Align reads to reference genome • assumed to be very similar, most reads will align • Identify sequence differences • SNPs, indels, rearrangements • Focus may be on • producing a catalogue of variants (1000 genomes) • producing a small number of very accurate genomes (mouse, Arabidopsis) • Generate new genome sequences
Mapping Accuracy in simulated human data Effects of Indels
Read Mapping:read length matters E.Coli 5.4 Mb S. cerevisiae 12.5 Mb A thaliana 120 Mb H sapiens 2.8 Gb 2008 18: 810-820 Genome Res.
Read Mapping(1): Hashing • Each nucleotide can be represented as a 2-bit binary number A=00, C=01, G=10, T=11 • A string of K nucleotides can be represented as a string of 2K bits eg AAGTC = 0000101101 • Each binary string can be interpreted as a unique integer • All DNA strings of length K can be mapped to the integers 0,1,…..4K-1 • k=10 65,535 • k=11 262,143 • k=12 1,048,575 • k=13 4,194,303 • k=14 16,777,215 • k=15 67,108,864 (effective limit for 32-bit 4-byte words) • Can use this relationship to index DNA for fast mapping • Need not use contiguous nucleotides – spaced seeds, templates • Trade-off between unique indexing/high memory use
MAQ Package for read mapping, SNP calling and management of read data and alignments Genome Research 2008 Easy to use - unix command-line based Although no longer state of the art and comparatively slow , generally produces good results
MAQ Read Mapping • Indexes all reads in memory and then scans through genome • Uses the first 28bp of each read for seed mapping • Guarantees to find seed hits with no more than two mismatches, and it also finds 57% of hits with three mismatches • Uses a combination of 6 hash tables that index different parts of each read to do this • Defines a PHRED-like read mapping quality • Qs = −10log10Pr{read is wrongly mapped}. • Based on summing the base-call PHRED scores at mismatched positions • Reads that map equally well to multiple loci are randomly assigned one location (and have Q=0) • Uses mate pair information to look for pairs of reads correctly oriented within a set distance • Defines mapping quality for a pair of consistent reads as the sum of their individual mapping qualities
Read-Mapping (2)Bowtie, BWA, Stampy all use the Burrows-Wheeler transform
Burrows Wheeler transform • Represents a sequence in a form such that • The original sequence can be recovered • Is more compressible (human genome fits into RAM) • similar substrings tend to occur together (fast to find words)
Bowtiehttp://bowtie-bio.sourceforge.net/index.shtmlhttp://genomebiology.com/2009/10/3/R25Bowtiehttp://bowtie-bio.sourceforge.net/index.shtmlhttp://genomebiology.com/2009/10/3/R25 • Uses the BWA algorithm • Indexes the genome, not the reads • Not quite guaranteed to find all matching positions with <= 2 mismatches in first 28 bases (Maq’s criterion) • Very fast (15-40 times faster than Maq) • Low memory usage (1.3 Gb for human genome) • Paper focuses on speed and # of mapped reads, not accuracy.“[…] Bowtie’s sensitivity […] is equal to SOAP’s and somewhat less than Maq’s”
Stampy (GertonLunter, WTCHG) • “Statistical Mapper in Python” (+ core in C) • Uses BWA and hashing • <= 3 mismatches in first 34 bp match guaranteedMore mismatches: gradual loss of sensitivity • Algorithm scans full read, rather than just beginning(and no length limit) • Handles indels well: Reads are aligned to reference at all candidate positions • Faster than Maq, slower than Bowtie • 2.7 Gb memory (shared between instances)
Performance – sensitivity Mapping sensitivity Indel size Top panel:Sensitivity for reads with indelsRight-hand panel:Sensitivity as function of divergence(Genome: human)
Viewing Read Alignments: lookseqhttp://www.sanger.ac.uk/resources/software/lookseq/
Viewing Read Alignments: IGVhttp://www.broadinstitute.org/igv/
Variant calling • A hard problem, several SNP callers exist eg MAQ/SAMTOOLS, Platypus (WTCHG) GATK (Broad) • Issue is to distinguish between sequencing errors and sequence variants • If variant has been seen before in other samples then problem is easier • genotyping vs variant discovery • VCF Variant call format is now standard file format • MAQ Assumes genome is diploid by default • Error model initially assumes that sequence positions are independent, attempts to compute probability of sequence variant • Has to use number of heuristics to deal with misalignments • SNP caller now part of SAMTOOLS varFilter.pl • acts as a filter on a large number of statistics tabulated about each sequence position
Problems with Variant Calling • Variant Calling is difficult because • a diploid genome will have two haplotypes present, which can differ significantly, eg due to polymorphic indels • should be easier with haploid or inbred genomes • but even harder when looking at low-coverage pools of individuals (eg 1000 genomes) • Coverage can vary depending on GC content • problem is sporadic • Optical duplicates may give the impression there is more support for a variant • often all reads with the same start and end points are thinned to a single representative, but this can cause problems if the coverage is very high • read misalignments can produce false positives • repetitive reads can be mapped to the wrong place • indels near the ends of reads can cause local read misalignments, where mismatches (SNPs) are favoured over indels • very divergent sequence is hard to align • may fail to give any mapping signal and will look like a deletion • problem addressed by local indel realignment (GATK)
GC content can affect read coverage (Arabidopsis data from Plant Sciences, thanks to Eric Belfield and Nick Harberd) coverage GC content • Possible causes: • Sanger identified that melting the gel slice by heating to 50 °C in chaotropic buffer decreased the representation of A+T-rich sequences. Nature methods | VOL.5 NO.12 | DECEMBER 2008 • PCR bias during library amplification. Nature methods | VOL.6 NO.4 | APRIL 2009 | 291 global
Deletions can cause SNP artefacts, by inducing misalignments at ends of reads Arabidopsis Data from Eric Belfield and Nick Harberd, Plant Sciences, Oxford
de-Novo genome assembly • No close reference genome available • Harder than resequencing • Only about 80-90% of genome is assembleable due to repeats • contiguation • scaffolding • Different Algorithms • More data required: • greater depth of coverage • range of paired-end insert sizes
Assembling Genomes from Scratchde-novo assembly • Software include: • VELVET • ABySS • ALL_PAIRS • SOAPdenovo • CORTEX
Computational limitations • Traditional approach to take reads as fundamental objects, and build algorithms/data structures to encode their overlaps • essentially quadratic in the number of reads • Next-generation sequencing machines generate too many reads! • simply holding the base-calls requires tens of terabytes for large projects • analysis produces lots of large intermediate files • Whatever we do, it has to scale slower than coverage