Download
1 / 39
390 likes | 524 Views
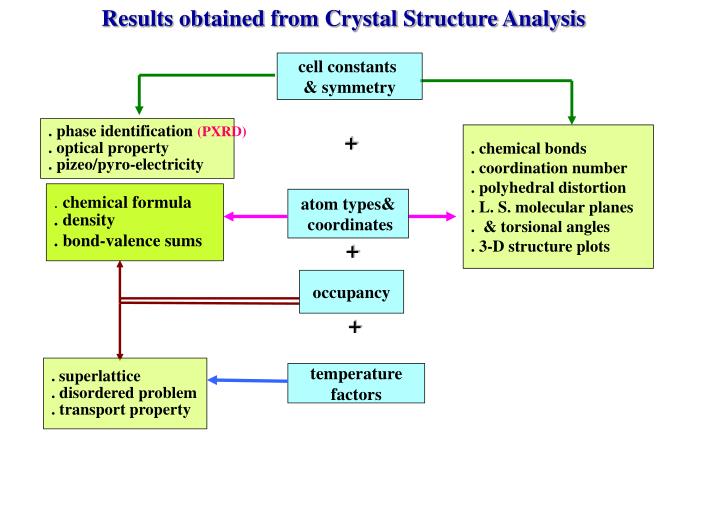
Results obtained from Crystal Structure Analysis. cell constants & symmetry. . phase identification (PXRD) . optical property . pizeo/pyro-electricity. +. . chemical formula . density . bond-valence sums. atom types& coordinates. +. occupancy. +. . superlattice

E N D
Results obtained from Crystal Structure Analysis cell constants & symmetry . phase identification (PXRD) . optical property . pizeo/pyro-electricity + . chemical formula . density . bond-valence sums atom types& coordinates + occupancy + .superlattice . disordered problem . transport property temperature factors . chemical bonds . coordination number . polyhedral distortion . L. S. molecular planes . & torsional angles . 3-D structure plots
Bond Valence Sum calculations Bond strength si= exp [(r0 - ri )/B] ri is an observed value;r0empirical value with B = 0.37 • Abond-valencecan be assigned toeach bond If the interatomic distances are known, the bond-valences can be calculated. BVS =isi • The sum of thebond-valences at each atom is equal to the magnitude of the atomic valence Examples: Determine the BVS for V, Ag and Na atoms.
R0 of some selected atoms Metal Ligand R0 Metal Ligand R0 V +3 O -2 1.743 V +4 O -2 1.784 V +5 O -2 1.803 Cr +2 O -2 1.73 Cr +3 O -2 1.724 Co +2 O -2 1.692 Co +3 O -2 1.70 Cu +1 O -2 1.610 Cu +2 O -2 1.679 Cu +3 O -2 1.739 Na +1 O -2 1.803 Rb +1 O -2 2.263 Cs +1 O -2 2.417 Ag +1 O -2 1.842 Be +2 O -2 1.381 Ca +2 O -2 1.967 Ba +2 O -2 2.285 Al +3 O -2 1.620 As +3 O -2 1.789 As +5 O -2 1.767
The relationship between coordination and valence of vanadium International Journal of Inorganic Materials 2 (2000) 561-579
International Journal of Inorganic Materials 2 (2000) 561-579
nce V5+ ? V4+ ? nce [V3O4(OH)(PO4)2]2-
BVS for V atoms [V3O4(OH)(PO4)2]2- (BVS) [(VVO2)(VIVO)2(OH)(PO4)2]2-
Ag-O: 2.394 ~ 4.015 Å The coordination number and BVS for Ag atom 2.394 (2x) 2.611 (2x) 2.659 (2x) 3.025 (2x) 3.382 (2x) 4.015 (2x)
BVS = 1.002 for C.N. = 8 2.394 (2x) 2.611 (2x) 2.659 (2x) 3.025 (2x) 3.382 (2x) 4.015 (2x) for C.N. = 10 BVS = si What will be the coordination number for Ag? Both BVS and the gap between bond lengths should be considered.
The coordination number and BVS for Rb atom BVS = 1.02 bond length gap
Atom x y z Ueq Na(1) 0.5932(1) 0.1926(1) -0.0298(2) 0.0191(5) Na(2) 1/3 -0.0528(2) -1/12 0.0251(8) Na(3) 0.3287(5) 0.1810(5) 0.044(1) 0.057(3)
The coordination number and BVS for Na atom C.N. = 6 C.N. = 5
Determination of chemical formula: What is the molecular formula of the organic component? (see ORTEP)
? Determination of chemical formula and coordination number for K atoms:
(1) Tables of crystal data, atomic coordinates, thermal parameters Crystallographic Data Journal: Inorg. Chemistry
what’s the chemical formula? Atomic coordinates and thermal parameters wrong if the atom is non-positive definite
A “bond” exists between two atoms A and B when DAB RA + RB + = 0.5Å (default value) inter-atomic distance ionic radii To look for H-bonds or other interatomic interactions beyond regular covalent or ionic bonds, can be set to larger values, say, 1 ~ 2 Å . tolerance
Least-square planes # MPLN: molecular plane
Torsional (or dihedral) angles The torsional (or dihedral) angle of four atoms A, B, C, D with a chemical bond between AB, BC and CD, is defined as the angle between the two planes through A, B, C, and B, C, D. The torsional angle is considered positive when it is measured clockwise from the front substituent A to the rear substituent D and negative when it is measured anti-clockwise.
l Structure factor table
(4) Respresentation of molecular and 3D structures ORTEP diagram Oak Ridge Thermal Ellipsoid Plot
(4) Respresentation of molecular and 3D structures Table S2. Atomic coordinates (x 104) and equivalent isotropic displacement parameters (Å2x 103) for NTHU-2. ___________________________________________________________x y z U(eq)a Zn(1) -495(1) 11778(1) -5962(1) 18(1) Zn(2) -4516(1) 11782(1) -5881(1) 24(1) P(1) -266(1) 10803(1) -3046(1) 18(1) P(2) -4766(1) 10775(1) -3011(1) 24(1) O(1) -326(1) 12031(3) -4062(3) 26(1) O(2) -217(1) 11431(3) -1613(3) 24(1) O(3) 163(1) 9848(4) -3285(4) 36(1) O(4) -737(1) 9809(4) -3126(4) 40(1) O(5) -4672(1) 11974(3) -4024(3) 28(1) O(6) -5242(1) 9981(4) -3266(4) 36(1) O(7) -4768(1) 11379(4) -1591(3) 33(1) O(8) -4352(1) 9596(4) -3111(4) 42(1) O(9) -1183(1) 11325(4) -6196(4) 37(1) O(10) -3819(1) 11657(4) -6160(4) 39(1) O(11) -1431(1) 13054(5) -7639(5) 51(1) O(12) -3565(1) 10408(5) -4341(5) 57(1) N(1) 4283(2) 14100(8) -5628(6) 60(2) N(2) 676(1) 13818(5) -5432(5) 37(1) C(1) -1503(2) 12044(6) -6842(5) 33(1) C(2) -2019(2) 11645(5) -6534(5) 28(1) C(3) -2400(2) 12320(7) -7279(6) 42(1) C(4) -2876(2) 12088(6) -6947(6) 40(1) C(5) -2980(2) 11191(6) -5859(5) 32(1) C(6) -2604(2) 10456(6) -5178(6) 35(1) C(7) -2126(2) 10700(6) -5502(5) 35(1) C(8) -3496(2) 11056(6) -5402(6) 34(1) C(9) 4230(2) 12996(10) -4822(9) 73(3) C(10) 3804(2) 12823(8) -4172(6) 50(2) C(11) 3434(2) 13815(6) -4348(6) 36(1) C(12) 3507(2) 14971(7) -5210(7) 50(2) C(13) 3936(3) 15094(9) -5851(8) 67(2) C(14) 2977(2) 13663(8) -3567(7) 51(2) C(15) 2509(2) 13809(7) -4395(7) 45(2) C(16) 2073(2) 13599(8) -3503(7) 50(2) C(17) 801(2) 12646(6) -4689(6) 42(1) C(18) 1254(2) 12562(6) -4101(7) 42(1) C(19) 1583(2) 13668(6) -4238(5) 31(1) C(20) 1450(2) 14882(6) -5039(6) 39(1) C(21) 984(2) 14938(6) -5622(6) 38(1) ________________________________________________________________________________ aU(eq) is defined as one third of the trace of the orthogonalized Uij tensor.
About the agreement factors About the intensity data (5) Justification of the crystal structure results • Is R1 (or RF) below 5%? If not, any rational explanation? • Is R1 close to Rint? • Is GOF (goodness-of-fit, or S) close to 1? •Is the resolution of the data collected below 0.9 Å? • Has absorption correcton been applied? • Are the criteria for “observed” data set properly?
(5) Justification of the crystal structure results About the results About the refinement •Has a proper weighting scheme been chosen? • Is the data-to-parameter ratio larger than 8? • Does the refinement converge without significant correlation? • Are thermal eliposids normal? • Has the absolute configuration been considered if acentric? • Can all H atoms be located on Fourier difference map? •Are the esds’in bond lengths smaller than 0.005 Å? • Are bond lengths and angles reasonable? • Do metal atoms possess proper coordinaton geometry? • Has the charge been balenced in the chemical formula? • ……...
Point-group symmetry and physical properties of crystals The point group of a crystal is a subgroup of the symmetry group of any of its physical properties. We can derive information about the symmetry of a crystal from its physical properties (Neumann’s principle) Certain interesting physical properties occur only in non-centrosymmetric crystals. EnantiomorphismEnantiomerism Chirality Dissymmetry These terms refer to the same symmetry restriction, the absence of improper rotations in a crystal or molecule
In particular, the absence of a center of symmetry, 1-bar, and of a mirror plane, m, but also of a 4-bar axis. As a consequence, such chiral crystals or molecules can occur in two different forms, which are related as a right and a left hand; hence they are called right-handed and left-handed forms. These two forms of a molecule or a crystal are mirror-related and not superimposable (not congruent). Thus the only symmetry operations which are allowed for chiral objects are proper rotations. Such objects are also calleddissymmetric, in contrast to asymmetric objects which have no symmetry. The terms enantiomerism and chirality are mainly used in chemistry and applied to molecules, whereas the term enantiomorphism is preferred in crystallography if reference is made to crystals.
About Oral presentation 1. Background of your crystalline sample species, color, size, stability, growth, …etc. 2. Justification of your intensity data 3. Justification of the assigned space group 4. How well is the first structure model? 5. The progression of your structure refinements 6. List a complete table of Crystal Data 7. List a complete table of atomic coordinates 8. List selected bond distances and angles 9. Prepare a CIF for your structure 10. Description of your structure ORTEP and 3D plots, geometric calculations, and structure features
Table A-1a. Crystal data and structure refinement for Na5InSi4O12. --------------------------------------------------------------------------------------------------------------------- Empirical formula InNa5O12Si4 Formula weight 534.13 Color; Habit colorless; rod Crystal size 0.05 x 0.05 x 0.15 mm3 Crystal system; space group Rhombohedral; R-3c Unit cell dime nsions a = 21.7158(9) Å c = 12.4479(7) Å Volume 5083.7(4) Å3 Z 18 Reflection for cell 4715 Density (calculated) 3.140 Mg/m3 Absorption coefficient 2.776 mm-1 F(000) 4608 Temperature 295 K Wavelength 0.71073 Å Theta range for data collection 1.88 to 28.29° Index ranges -28 ≤ h ≤ 28, -28 ≤ k ≤ 28, -16 ≤ l ≤ 7 Reflections collected 11968 Independent reflections 1415 (1363 2 (I)) [R(int) = 0.0755] Completeness to theta = 28.29° 100.0 % Absorption correction semiempirical (based on 1815 reflections) Max. and min. transmission 0.968 and 0.860 Refinement method Full-matrix least-squares on F2 Data / restraints / parameters 1415 / 0 / 111 Goodness-of-fit on F2 1.574 Final R indices [I>2sigma(I)] R1a = 0.0452, wR2b = 0.0947 R indices (all data) R1 = 0.0468, wR2 = 0.0951 Largest diff. peak and hole 0.621 and -1.072 e∙Å-3 aR1 = Σ||FO|-|FC|| / Σ| FO | bwR2 = [Σ w(FO 2-FC 2)2 / Σ w(FO 2)2]1/2, w =1 / [2(FO2) + ( 0.0147P )2 + 82.37P] where P =