Download

1 / 128
1.28k likes | 1.41k Views
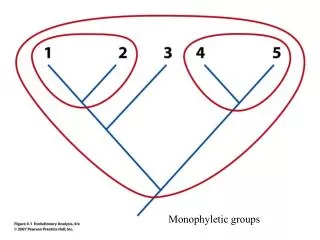
GLOBAL PAIRWISE ALIGNMENT GLOBAL ALIGNMENT OF: 2 NUCLEOTIDE SEQUENCES OR 2 AMINO-ACID SEQUENCES. Assumptions: Life is monophyletic Biological entities (sequences, taxa) share common ancestry. ancestor. descendant 1. descendant 2. Any two organisms share a common ancestor in their past.

E N D
GLOBAL PAIRWISE ALIGNMENTGLOBAL ALIGNMENT OF: 2 NUCLEOTIDE SEQUENCES OR 2 AMINO-ACID SEQUENCES
Assumptions: Life is monophyletic Biological entities (sequences, taxa) share common ancestry
ancestor descendant 1 descendant 2 Any two organisms share a common ancestor in their past
(~5 MYA) ancestor
(~120 MYA) ancestor
(~1,500 MYA) ancestor
(1) Speciation events (2) Gene duplication (3) Duplicative transposition Homologous sequences
Homology:A term coined by Richard Owen in 1843. Definition: Similarity resulting from common ancestry.
Homology There are three main types of molecular homology: orthology, paralogy (including ohnology) and xenology.
Homology: General Definition • Homology designates a qualitative relationship of common descent between entities • Two genes are either homologous or they are not! • it doesn’t make sense to say “two genes are 43% homologous.” • it doesn’t make sense to say “Linda is 43% pregnant.”
Orthology & Paralogy • Two genes are orthologs if they originated from a single ancestral gene in the most recent common ancestor of their respective genomes • Two genes are paralogs if they are related by gene duplication. Two genes are ohnologs if they are related by gene duplication due to genome duplication
Xenology is due to horizontal (lateral) gene transfer (HGT or LGT) XA and XB are xenologs Distinguishing orthologs from xenologs is impossible in pairwise genomic comparisons, but possible when multiple genomes are compared
Orthology, Paralogy, Xenology(Fitch, Trends in Genetics, 2000. 16(5):227-231)
Homology By comparing homologous characters, we can reconstruct the evolutionary events that have led to the formation of the extant sequences from the common ancestor.
Homology When comparing sequences, we are interested in POSITIONAL HOMOLOGY. We identify POSITIONAL HOMOLOGY through SEQUENCEALIGNMENT.
Alignment: A hypothesis concerning positionalhomology among residues from two or more sequence. Positional homology = In pairwise alignment, a pair of nucleotides from two homologous sequences that have descended from one nucleotide in the ancestor of the two sequences.
Sequence alignment involves the identification of the correct location of deletions and insertions that have occurred in either of the two lineages since their divergence from a common ancestor.
Unknown sequence Unknown events & unknown sequence of events Unknown events & unknown sequence of events The true alignment is unknown.
There are two modes of alignment. Global alignment: each residue of sequence A is compared with each residue in sequence B. Global alignment algorithms are used in comparative and evolutionary studies. Local alignment: Determining if sub-segments of one sequence are present in another. Local alignment methods have their greatest utility in database searching and retrieval (e.g., BLAST).
For reasons of computational complexity, sequence alignment is divided into two categories: Pairwise alignment (i.e., the alignment of two sequences). Multiple-sequence alignment (i.e., the alignment of three or more sequences). Pairwise alignment problems have exact solutions. Multiple-sequence alignment problems only have approximate (heuristic) solutions.
A pairwise alignment consists of a series of paired bases, one base from each sequence. There are three types of pairs:(1) matches = the same nucleotide appears in both sequences. (2) mismatches = different nucleotides are found in the two sequences. (3) gaps = a base in one sequence and a null base in the other. GCGGCCCATCAGGTAGTTGGTG-G GCGTTCCATC--CTGGTTGGTGTG
-Two DNA sequences: A and B.-Lengths are m and n, respectively. -The number of matched pairs is x. -The number of mismatched pairs is y. - Total number of bases in gaps is z.
There are internal and terminal gaps. GCGG-CCATCAGGTAGTTGGTG-- GCGTTCCATC--CTGGTTGGTGTG
A terminal gap may indicate missing data. GCGG-CCATCAGGTAGTTGGTG-- GCGTTCCATC--CTGGTTGGTGTG
An internal gap indicates that a deletion or an insertion has occurred in one of the two lineages. GCGG-CCATCAGGTAGTTGGTG-- GCGTTCCATC--CTGGTTGGTGTG
When sequences are compared through alignment, it is impossible to tell whether a deletion has occurred in one sequence or an insertion has occurred in the other. Thus, deletions and insertions are collectively referred to as indels (short for insertion or deletion). GCGG-CCATCAGGTAGTTGGTG-- GCGTTCCATC--CTGGTTGGTGTG
The alignment is the first step in many functional and evolutionary studies. Errors in alignment tend to amplify in later stages of the study.
Motivation for sequence alignment Function • Similarity may be indicative of similar function. Evolution • Similarity may be indicative of common ancestry.
Methods of alignment: 1. Manual 2. Dot matrix 3. Distance Matrix 4. Combined(Distance + Manual)
Manual alignment.When there are few gaps and the two sequences are not too different from each other, a reasonable alignment can be obtained by visual inspection. GCG-TCCATCAGGTAGTTGGTGTG GCGATCCATCAGGTGGTTGGTGTG
Advantages of manual alignment: (1) use of a powerful and trainable tool (the brain, well… some brains).(2) ability to integrate additional data, e.g., domain structure, biological function.
Protein Alignment may be guided by Secondary and Tertiary Structures Escherichia coli DjlA protein Homo sapiens DjlA protein
Disadvantages of manual alignment:subjectivity (the algorithm is unspecified) irreproducibility (the results cannot be independently reproduced) unscalability(inapplicable to long sequences)incommensurability (the results cannot be compared to those obtained by other methods)
The dot-matrix method (Gibbs and McIntyre, 1970): The two sequences are written out as column and row headings of a two-dimensional matrix. A dot is put in the dot-matrix plot at a position where the nucleotides in the two sequences are identical.
The alignment is defined by a path from the upper-left element to the lower-right element.
There are 4 possible steps in the path: (1) a diagonal step through a dot = match. (2) a diagonal step through an empty element of the matrix = mismatch. (3) a horizontal step = a gap in the sequence on the left of the matrix. (4) a vertical step = a gap in the sequence on the top of the matrix.
A dot matrix may become cluttered. With DNA sequences, ~25% of the elements will be occupied by dots by chance alone.
window size =1 stringency = 1 alphabet size = 4 The number of spurious matches is determined by: window size(how many residues are compared), stringency (the minimum number of matches for a hit), & alphabet size (number of characters states). Window size must be an odd number.
window size =1 stringency = 1 alphabet size = 4 window size = 3 stringency = 2 alphabet size = 4
window size = 1 stringency = 1 alphabet size = 20
Dot-matrix methods:Advantages: By being a visual representation, and humans being visual animals, the method may unravel information on the evolution of sequences that cannot easily be gleaned from a line alignment.Disadvantages: May not identify the best possible alignment.
The vertical gap indicates that a coding region corresponding to ~75 amino acids has either been deleted from the human gene or inserted into the bacterial gene. Window size = 60 amino acids; Stringency = 24 matches Advantages: Highlighting Information
Window size = 60 amino acids; Stringency = 24 matches Advantages: Highlighting Information The two pairs of diagonally oriented parallel lines most probably indicate that two small internal duplications occurred in the bacterial gene.
Disadvantages: Not possible to identify the best alignment.