Download

1 / 23
240 likes | 1.45k Views
CPAM: Congenital Pulmonary Airway Malformation CCAM Congenital Cystic Adenomatoid Malformation. Kristina Reitz Néonatologie - 1.6.2010. 1. Yuma. Suspicion anténatale de malformation adenomatoïde kystique du poumon droit à l’échographie de 19 SA, confirmée aux échographies suivantes.

E N D
CPAM: CongenitalPulmonaryAirway MalformationCCAMCongenitalCysticAdenomatoid Malformation Kristina Reitz Néonatologie - 1.6.2010
1. Yuma • Suspicion anténatale de malformation adenomatoïde kystique du poumon droit à l’échographie de 19 SA, confirmée aux échographies suivantes. • Naissance à 40+4/7 SA par césarienne (siège), bonne adaptation, Apgar 9/10/10, PN 3 kg. • Aucune symptomatologie respiratoire – pas de dyspnée ni tachypnée, pas de toux. • s’alimente bien au sein
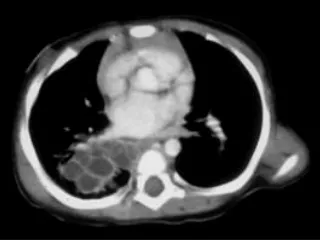
Rx initiale ???
Rx initiale « Image radiotransparente hétérogène localisée au niveau de l’apex du lobe inférieur droit, compatible avec une malformation adénomatoïde kystique. »
Yuma - évolution • 1er CT thoracique à J10: malformation adenomatoïde kystique du lobe inférieur droit de type 2 • contrôles cliniques mensuels, asymptomatique, bonne croissance • 2è CT (injecté) vers 6-8 mois: au vu de l’étendue des lésions, une régression spontanée complète semble peu probable. • Opération : Lobectomie inférieure et moyenne par thoracotomie droite à 10 mois, compliquée d’un pneumothorax nécessitant un drainage, puis évolution favorable
Yuma: CT injecté à 7 mois • On retrouve les deux lésions lobaires inférieures droites, l'une apicale, l'autre basale, composées de multiples kystes dont les plus grands mesurent environ 6 à 7mm de diamètre, en augmentation de taille comparativement à l'examen précédent. La lésion apicale mesure environ 48 x 34mm de grand diamètre (34 x 22mm à l'examen précédent) et la lésion basale mesure environ 34 x 31mm (24 x 14mm à l'examen précédent). • La croissance du parenchyme pulmonaire semble relativement proportionnelle compte tenu de l'intervalle entre les examens. • Pas d'évidence de pédicule vasculaire au niveau des deux lésions, pas d'autre anomalie suspecte loco-régionalement, les lésions sont compatibles avec des lésions de type CCAM 2.
2. Noé, 6 ans • Diagnostic anténatal de malformation adenomatoïde kystique de type 1 du LIG et suspectée au LID sur l’IRM anténatale • In-utéro, kystes ponctionnés et drainés à plusieurs reprises. L’ultrason montre un kyste dans le poumon gauche de 5 cm de diamètre, avec un refoulement du parenchyme pulmonaire vers le haut et du médiastin vers la droite. • Accouchement à 39 +6/7 par césarienne en semi-urgence avec procédure EXIT : le bébé reste en circulation fœtale, rattaché au cordon, et on procède à une nouvelle ponction du kyste. Immédiatement après, on clampe le cordon, le bébé est intubé et mis sous ventilation HFO
Noé • Lobectomie du LIG à J2. • Pas de CCAM au LID. • HTAP d’évolution favorable par la suite. • Extubé après 6 semaines, persistance d’une O2-dépendance • A l’âge de 2 mois, RAD sous O2, suivi en pneumologie. • Oxygénothérapie à domicile avec besoin d’O2 nocturne jusqu’à l’âge de 4 ans ainsi qu’en cas d’exacerbation respiratoire. • Infections des voies respiratoires à répétition avec multiples hospitalisations; mise sous antibiothérapie alternée pendant les mois d’hiver
Noé: bilan pneumo à 5 ans • CT: Déformation thoracique sévère avec pectusexcavatum et shift médiastinal à droite. Hypoplasie pulmonaire droite et hyperinflation modérée à gauche associées à des tractus fibreux postéro-basaux gauches. • Scintigraphie de perfusion pulmonaire: asymétrie de taille et de fonction en défaveur du poumon gauche qui représente 35% de la fonction pulmonaire totale. Une étude de la ventilation n’a pas été possible au vu du jeune âge. • Bronchoscopie: Malacie de la bronche souche G • Pas d’HTAP. • scintigraphie de ventilation/perfusion ainsi que des fonctions pulmonaires pas encore possibles (collaboration).
CPAM • Malformation pulmonaire kystique la +fréquente • Rare: env. 1/8000 – 1/35000 naissances • ↑ diagnostic depuis introduction de l’écho anténatale • En général limitée à un lobe, 10% affectant plusieurs lobes • Sporadique, pas de facteurs de risque ni d’origine génétique connus
Pathologie • Lésions hamartomateuses de l’arbre bronchique vs. arrêt localisé du dvt de l’arbre bronchique fétal • Différents types: différentes origines spatio-temporelles? • Pathogénèse: imbalance entre prolifération cellulaire et apoptose? GDNF (glial derivedneurotrophic factor)? • possible compression du tissu pulmonaire adjacent entrainant une hypoplasie pulmonaire • CPAM/CCAM: connecté à l’arbre bronchique, vascularisation artérielle et veineuse par circulation bronchique
Type 1: « grands kystes » • CPAM la plus fréquente (65%) • Origine: bronches distales/bronchioles proximales • Tissu différencié – origine vers 7-10 SA • Grands kystes de 2-10 cm, « single » ou « multiloculated », remplis d’air ou de liquide, +/- mucus +/- cartilage • souvent compression des alvéoles adjacentes +/- déviation médiastinale
Type 2: « petits kystes » • Fréquence: 20-25% des CCAM • Kystes multiples 0,5 – 2cm avec zones plus solides, ressemblant à des bronchioles terminales dilatées • En général, pas d’effet de masse sur poumon adjacent • Associé à autres anomalies congénitales (-> 60%): atrésie de l’œsophage avec fistule, agénésie rénale, autres malformations pulmonaires, anomalies du diaphragme, du SNC et des os • Origine: 3 SA ?
Type 3: Type adénomatoïde • Fréquence: 8% des CCAM • Origine acinaire; vers 4-5 SA • Souvent très étendue, affectant un lobe entier ou plusieurs lobes • Petits kystes <0,5cm • Structure solide +/- kystique, manque de différenciation
Type 0 et type 4 • Tous les deux caractérisés par épithelium acinaire-alvéolaire ≠ épithélium bronchiolaire dans types 1-3 • Type 0: rare (1-3%), très petits kystes <0.5cm, +mucus +cartilage –muscle; souvent létal; souvent associé à des anomalies cardio-vasculaires • Type 4: rare (2-4% des CCAM), kystes de 7cm
Diagnostic anténatal • US/IRM: lésion petite à massive • Régression dans env. 60% des cas avec parfois résolution complète en postnatal • Hydropsfétal dans 40% (sur kystes larges et shift médiastinal -> compression de la V. cave et du cœur) +/- Polyhydramnios • Taille maximale des kystes vers 25 SA avec régression en général vers 30 SA si pas d’hydrops • Présentation très variable, asymptomatique ou détresse respiratoire néonatale
Présentation néonatale • Symptômes en période postnatale: 2/3 des CPAM symptomatiques • NN avec SDR, tachypnée, grunting, tirage et cyanose (souvent type1), +/- air trapping et expansion des grands kystes avec péjoration du SDR; évtl pneumothorax • Type 2: souvent reconnu en raison des malformations associées; SDR semblable au type1 • Type 3: le plus sévère; hydrops, hypoplasie pulmonaire, « stillborn » ou SDR sévère
Présentation tardive • En général, lésions plus petites • Pneumonies récurrentes ou toux, dyspnée, +/- cyanose • Plus rare: pneumothorax, hémoptysies • Examen clinique: asymétrie thoracique, hyperrésonnance, diminution des bruits à l’auscultation
DD • Pneumatocèles post-pneumonie • Emphysème pulmonaire interstitiel • Kyste pulmonaire simple • Kyste bronchogénique ayant une communication avec une bronche • Blastome pleuro-pulmonaire kystique
Risque de dégérescence? • CPAM type1: risque de carcinome bronchiolo-alvéolaire à l’âge adulte • Blastome pleuro-pulmonaire: lésion kystique maligne du jeune enfant, avec risque de dégénérescence en sarcome vers 2-6 ans • Clinique et Rx comme CPAM • Prédisposition génétique
Attitude thérapeutique • Taille de la lésion • Tttfétal: shuntthoraco-amniotiqueouthoraco-abdominal • Chirurgie: lobectomie (résection segmentale difficile), duréed‘hosp de 3-5j; lobectomie par thoracoscopie … • Ttt des enfantsasymptomatiques??? • Genève: à 6-8 mois, l’intervention est bien tolérée par l’enfant, et permet encore le développement et le rattrapage de la croissance pulmonaire et du nombre des alvéoles après lobectomie. • Suivi jusqu’à l’âge adulte
Réf • Up-to-date • Taussig et al.: PediatricRespiratoryMedicine 2008 • Congenitalcystadenomatoidmalformations: resectsomeandobserve all? Fitzgerald, PaedRespRev 2007 • PulmonaryCysts in earlychildhoodandtheriskofmalignancy. Priest et al., PedPulmonology 2009