Download
1 / 20
200 likes | 324 Views
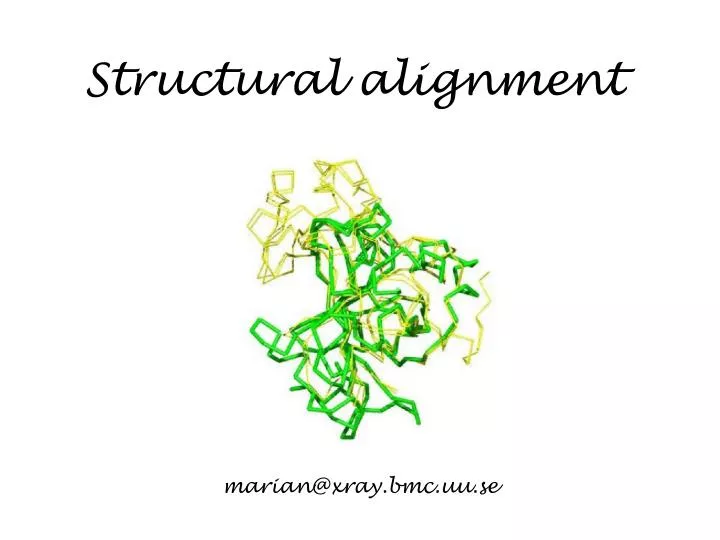
Structural alignment. marian@xray.bmc.uu.se. Protein structure. Every protein is defined by a unique sequence (primary structure) that folds into a unique shape (tertiary or three-dimensional structure). However, proteins with similar sequences adopt very similar structures.

E N D
Structural alignment marian@xray.bmc.uu.se
Protein structure Every protein is defined by a unique sequence (primary structure) that folds into a unique shape (tertiary or three-dimensional structure). However, proteins with similar sequences adopt very similar structures. Cyclophilin from B. malayi Cyclophilin A from H. sapiens
Why structural alignment ? we have sequence alignment - Clustal… KTHLCV KSHA -V that gives us an idea about a correspondence of amino acids of two (or more ) proteins That enables to infer information about function And evolution of the Protein If the sequences are similar enough !!!!
What is twilight zone ? Sequence alignment unambiguously distinguish only between protein pairs of similar structure and non-similar structures when the pairwise sequence identity is high. High sequence identity roughly means over 40 %. The signal gets blurred in the twilight zone of 20-35 % sequence identity.
More of the twilight zone More than 90 % sequence pairs with the sequence identity lower than 25 % have different structures. Significance of sequence alignments is length dependent. The longer the sequence the lower identity is required to be be called significant.Nevertheless, it converges to 25% with alignments longer than 80 amino acids. ‘The more similar than identical’ rule can reduce a number of false positives. Using of intermediate sequences for finding links between more distant families can also reduce a number of false positives.
How far can the sequence identity drop? Average sequence identity of random alignments - 5.6 % Average sequence identity of remote homologues 8.5 %
How does it work? From http://www.biochem.unizh.ch/antibody/Introduction/Institutsseminar97/source/slide2.htm
Numbers Given the average length of a protein 300 amino acid, there are 20300 possibilities of building the average protein - more than atoms in universe. In reality just few hundred thousand sequences are known. It is believed that a number of basic protein folds is between 1500 - 5000.
Structural alignment because: Structures are better conserved than sequences structural alignment can imply a functional similarity that is not detectable from a sequence alignment . Might help to improve sequence alignment when structures are available (phylogenetic studies, homology modeling). Will improve sequence alignment methods (use of structural alignments’ substitution matrices, gap penalties). Will improve sequence prediction methods
Sequence versus structural alignment 2 3 4 5 6 7 8 9 10 11 12 13 14 PHE ASP ILE CYS ARG LEU PRO GLY SER ALA GLU ALA VAL CYS PHE ASN VAL CYS ARG THR PRO --- --- --- GLU ALA ILE CYS PHE ASN VAL CYS ARG --- --- --- THR PRO GLU ALA ILE CYS
Is it difficult to make structural alignment? Structural alignment is NP-hard (nondeterministic polynomial time) problem. In other words, it is not tractable properly. Even, if it would, the result would be correct from technical point of view not necessary from biological point of view. Yes, it is.
General solution Use a heuristic approach: Represent the proteins A and B in some coordinate independent space Compare A and B Optimize the alignment between A and B (e.g. minimize R.M.S.d.) Measure the statistical significance of the alignment against some random set of structure comparisons
“..in some coordinate independent space…” • Make the problem easier by: • comparing only distance matrices of atoms • comparing secondary • structure element (SSE) • comparing cartoons • comparing vectors of SSE • combination of mentioned methods • ….
None of the methods guarantee the finding of the closest structure and two methods can disagree at all amino acid positions. Nevertheless they can still provide a valuable insight into the history of the protein and give hints concerning the function.
Protein structure classification If you want to know which structures are similar to a known structure, these systems might help: Manual - SCOP Semi-automatic - CATH Automatic - FSSP
CATH C (class) - secondary structure composition A (architecture) - overall shape, secondary structure elements orientation T (topology) - overall shape, secondary structure elements orientation + connectivity H (homologous superfamily) - Sequence identity >= 35%, 60% of larger structure equivalent to smaller SSAP score >= 80.0 and sequence identity >= 20% 60% of larger structure equivalent to smaller SSAP score >= 80.0, 60% of larger structure equivalent to smaller and domains which have related functions S (sequence families) - clustering based on the sequence identity level
Summary Structural alignment can help with protein annotations even when the sequence similarity is not significant. Sequence identity of two proteins with similar structures can be lower than 10 % - number of folds is limited. Recent progress in the protein structure determination increases the usefulness of structural alignment. Structural alignment is difficult problem that is solved by heuristic methods. These methods simplify the problem by moving from 3D space to 2D space sacrificing the optimum result for the speed.
Summary II Different methods can provide completely different alignments. In our results, CE, Dali,Matras and Vast were the best servers for finding structural relatives. A few structural classification systems were developed (CATH, FSSP, SCOP), they provide hierarchical classification of protein structures and enable to infer functional and evolutional relationships between proteins.