Download

1 / 26
260 likes | 416 Views
Tools used for genome and exome data processing.

E N D
Genome and Exome Data Processing- How to obtain high quality variant calls (mapping tools, variant calling tools and GATK best practices)- Variant reduction Center for Computational Molecular Biology, Brown University Alireza K. Jamayran 24th June, 2013
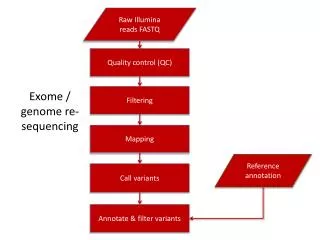
An Overview of Data Processing Raw reads Mapping ATTACGCATCGATCGGGTACCTTATTCGATCCGATT Variant calling
Common Raw Data (reads) QC and Manipulation Tools • Basic Statistics • Per base sequence quality • Per base sequence scores • Per base sequence content • Per base GC content • Per base sequence GC content • Per base N content • Sequence Length Distribution • Sequence Duplication Levels • Overrepresented sequences • EMBOSS • FastQC • BioPython • BioRuby • BioPerl • FASTX-Toolkit • BioJava • FASTQ-to-FASTA converter • FASTQ Information • FASTQ/A Collapser • FASTQ/A Trimmer • FASTQ/A Renamer • FASTQ/A Clipper • FASTQ Quality Box plot Graph • FASTQ/A Reverse-Complement • FASTQ/A Barcode splitter • FASTA Formatter • FASTA Nucleotide Changer • FASTQ Quality Filter • FASTQ Quality Trimmer • FASTQ Masker • FASTQ /A Quality Stats
Common Read Aligner/Mapping Tools Read Seq. hashing Genome hashing String matching using BWT • SeqMap • SHRiMP • ZOOM • RMAP • MAQ • CloudBurst • SOAP v1 • Mosaik • Corona Lite • SSAHA2 • NovoAlign • ProbeMatch • PASS • SMALT • ReSEQ • Bfast • Bowtie • BWA • SOAP v2 Heng Li and Richard Durbin, Fast and accurate short read alignment with Burrows–Wheeler transform. BIOINFORMATICS, Vol. 25 no. 14 2009, pages 1754–1760
Visualization of Mapped Data Integrative Genomics Viewer (IGV) Integrated Genome Browser (IGB) Tview (samtools) UCSC Genome Browser
Common Variant Calling Tools • Mosaik –Mapping (Illumina,454, SOLID) – Variant calling • SOAP – Mapping (Illumina) – SNP calling • Samtools – Data processing – Variant calling • GLFtools – SNP calling • QCALL – SNP calling • GATK – Data processing – Variant calling – Variant manipulation – Variant QC • DINDEL – Indel calling
1000 Genomes Project • Initial phase (pilot project) • Phase I : 1092 samples • Phase II : about 1700 samples • Phase III : 2500 samples (Alignment data release MAY 25, 2013)
Tools Used for 1000 Genomes Pilot Project Data Processing Trio and low coverage pilots Exon pilot 454 data Illumina data 454 data SOLiD data Illumina data Mapping MAQ Corona Lite SSAHA2 MOSAIK All data from Illumina and 454 platforms has been recalibrated using the theGATK package. Recalibration SNP calling Multiple SNP calling procedures have been used. This was achieved using the IMPUTE2 software (Howie, Donnelly et al. 2009) to produce best-guess haplotypes from unphased genotype data. Phasing Indel calling Dindelwas used for indel calling. For the main project BWA is used for Illumina and BFAST for SOLiD, 454 started with SSAHA but the final set of 454 alignments used SMALT
1000 Genomes04-2012 release (VCF file converted to tab-delimited) • AMR 20,664,411 • ASN 15,048,709 • AFR 26,708,385 • EUR 17,622,236 • ALL (04-2012)39,706,714 • ALL (02-2012) 42,074,823 • What is the lowest allele frequency in 1000 genomes?
Exome Server Project (ESP6500/ESP5400) • Mapping • BWA was used for mapping. PCR Duplicates were removed using Picard. Alignments were recalibrated using GATK. • SNP calling • SNPs were called using a two-step approach. First, genotype likelihood files (GLFs) were generated usingsamtoolspileup on individual BAM files. Next, glfMultipleswas used, a multi-sample variant caller, to generate initial SNP calls • Indel calling • Small INDEL variants were analyzed at the Broad Institute (by the Genome Sequencing and Analysis group) using the GATK.
A Typical Data Processing Workflow at NDAL (Neuro-degeneration Research Lab., Bogazici University)
Duplicate Marking Before Duplicate Marking ACGGTACGGGTAACGTTGCTGACTGCATTCAG Duplicates After Duplicate Marking ACGGTACGGGTAACGTTGCTGACTGCATTCAG
Realignment (Indels in the file and indels in the training file)
Base Quality Score Recalibration (BQSR) (by training your data) • Quality scores generated by sequencing machines are biased. • Inaccurate quality scores are abundant specifically at the end of the reads (due to machine cycling). • Proceeding and current nucleotide can bias the scores (due to sequencing chemistry effect).
Reducing Reads Before reducing reads ATTACGCATCGATCGGGTACCTTATTCGATCCGATT After reducing reads ATTACGCATCGATCGGGTACCTTATTCGATCCGATT
Variant Quality Score Recalibration (VQSR) • Most variant calling tools call a lot of false positives variants. • Hand-tuned filtering is hard and needs expertise ( you should know what filters to apply for each data) • VQSR learns from the data that what filters should be applied (AC=2;AF=1.00;AN=2;DP=10;HaplotypeScore=0.0000;MLEAC=2;MLEAF=1.00;MQ=58.11;MQ0=0;QD=30.88;VQSLOD=5.81) • You can train your data with known sites (HapMap, dbSNP, 1000G)
Phasing Before Phasing After Phasing
Common variant annotation tools • ANNOVAR • SeattleSeq • SnpEff • Genome Trax
Common Prediction Tools • AVSIF • SIFT • PolyPhen 2 • HGMD • LRT • Mutation Taster Consequence Prediction • PhastCons • PhyloP • Genomic Evolutionary Rate Profiling (GERP) Conservation Prediction
Things to Consider for Variant Reduction • Prevalence of a disease • The genetics of a disease • Symptoms • Population structure • Family structure (pedigree)
dbSNP • Common/non-flagged SNPs - SNPs with >= 1% minor allele frequency (MAF), mapping only once to reference assembly. • Flagged SNPs- SNPs < 1% minor allele frequency (MAF) (or unknown), mapping only once to reference assembly, flagged in dbSnp as "clinically associated" -- not necessarily a risk allele! • Mult. SNPs- SNPs mapping in more than one place on reference assembly. • All SNPs - all SNPs from dbSNP mapping to reference assembly.