Download

1 / 27
310 likes | 705 Views
Genome & Exome Sequencing Read Mapping. Xiaole Shirley Liu STAT115, STAT215, BIO298, BIST520. Whole Genome Sequencing. Usually need 30-50X coverage (~ 3 lanes of 100bp PE HiSeq2000 sequencing). Exome Sequencing. 2011. Exome Sequencing.

E N D
Genome & Exome SequencingRead Mapping Xiaole Shirley Liu STAT115, STAT215, BIO298, BIST520
Whole Genome Sequencing • Usually need 30-50X coverage (~ 3 lanes of 100bp PE HiSeq2000 sequencing)
Exome Sequencing • 2011
Exome Sequencing • Solution Hybrid Selection: Probes in solution can capture all exons (exome) for high throughput sequencing • 1-2% of whole genome seq • Easily multiplex 20 samples in one lane
Comparative Sequencing • Somatic mutation detection between normal / cancer pairs • WGS or WES • More mutation yield and better causal gene identification than Mendelian disorders Meyerson et al, Nat Rev Genet 2010
Hallmark of Mendelian Disease Gene Discovery Gilissen, Genome Biol 2011
Hallmark of Mendelian Disease Gene Discovery Gilissen, Genome Biol 2011
Mutation Targets vs Disorder Frequency Rarer disorders are focused on fewer mutated genes Gilissen, Genome Biol 2011
Whole Genome or Exome Seq? • Enabling technologies: NGS machines, open-source algorithms, capture reagents, lowering cost, big sample collections • Exomes more cost effective: Sequence patient DNA and filter common SNPs; compare parents child trios; compare paired normal cancer • Challenges: • Still can’t interpret many Mendelian disorders • Rare variants need large samples sizes • Exome might miss region (e.g. novel non-coding genes) • Unsuccessful at using exome-seq to interpret clinical data Shendure, Genome Biol 2011
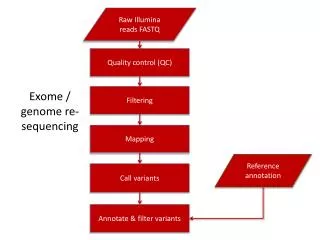
Read Mapping • Mapping hundreds of millions of reads back to the reference genome is CPU and RAM intensive, and slow • Read quality decreases with length (small single nucleotide mismatches or indels) • Very few mapper deals with indel, and often allow ~2 mismatches within first 30bp (4 ^ 28 could still uniquely identify most 30bp sequences in a 3GB genome) • Mapping output: SAM (BAM) or BED
Spaced seed alignment • Tags and tag-sized pieces of reference are cut into small “seeds.” • Pairs of spaced seeds are stored in an index. • Look up spaced seeds for each tag. • For each “hit,” confirm the remaining positions. • Report results to the user.
Burrows-Wheeler • Store entire reference genome. • Align tag base by base from the end. • When tag is traversed, all active locations are reported. • If no match is found, then back up and try a substitution. Trapnell & Salzberg, Nat Biotech 2009
Burrows-Wheeler Transform • Reversible permutation used originally in compression • Once BWT(T) is built, all else shown here is discarded • Matrix will be shown for illustration only T BWT(T) Burrows Wheeler Matrix Last column Burrows M, Wheeler DJ: A block sorting lossless data compression algorithm. Digital Equipment Corporation, Palo Alto, CA 1994, Technical Report 124; 1994 Slides from Ben Langmead
Burrows-Wheeler Transform • Property that makes BWT(T) reversible is “LF Mapping” • ith occurrence of a character in Last column is same text occurrence as the ith occurrence in Firstcolumn Rank: 2 BWT(T) T Rank: 2 Burrows Wheeler Matrix Slides from Ben Langmead
Burrows-Wheeler Transform • To recreate T from BWT(T), repeatedly apply rule: T = BWT[ LF(i) ] + T; i = LF(i) • Where LF(i) maps row i to row whose first character corresponds to i’s last per LF Mapping FinalT Slides from Ben Langmead
Exact Matching with FM Index • To match Q in T using BWT(T), repeatedly apply rule: top =LF(top, qc); bot = LF(bot, qc) • Whereqc is the next character in Q (right-to-left) and LF(i, qc) maps row i to the row whose first character corresponds to i’s last character as if it were qc Slides from Ben Langmead
Exact Matching with FM Index • In progressive rounds, top & bot delimit the range of rows beginning with progressively longer suffixes of Q Slides from Ben Langmead
Exact Matching with FM Index • If range becomes empty (top = bot) the query suffix (and therefore the query) does not occur in the text Slides from Ben Langmead
Backtracking • Consider an attempt to find Q = “agc” in T = “acaacg”: • Instead of giving up, try to “backtrack” to a previous position and try a different base (much slower) • For 50bp reads, need to have ~25bp perfect match “g” “c” “gc” does not occur in the text Slides from Ben Langmead
Seq Files @HWI-EAS305:1:1:1:991#0/1 GCTGGAGGTTCAGGCTGGCCGGATTTAAACGTAT +HWI-EAS305:1:1:1:991#0/1 MVXUWVRKTWWULRQQMMWWBBBBBBBBBBBBBB @HWI-EAS305:1:1:1:201#0/1 AAGACAAAGATGTGCTTTCTAAATCTGCACTAAT +HWI-EAS305:1:1:1:201#0/1 PXX[[[[XTXYXTTWYYY[XXWWW[TMTVXWBBB HWUSI-EAS366_0112:6:1:1298:18828#0/1 16 chr9 98116600 255 38M * 0 0 TACAATATGTCTTTATTTGAGATATGGATTTTAGGCCG Y\]bc^dab\[_UU`^`LbTUT\ccLbbYaY`cWLYW^ XA:i:1 MD:Z:3C30T3 NM:i:2 HWUSI-EAS366_0112:6:1:1257:18819#0/1 4 * 0 0 * * 0 0 AGACCACATGAAGCTCAAGAAGAAGGAAGACAAAAGTG ece^dddT\cT^c`a`ccdK\c^^__]Yb\_cKS^_W\ XM:i:1 HWUSI-EAS366_0112:6:1:1315:19529#0/1 16 chr9 102610263 255 38M * 0 0 GCACTCAAGGGTACAGGAAAAGGGTCAGAAGTGTGGCC ^c_Yc\Lcb`bbYdTa\dd\`dda`cdd\Y\ddd^cT` XA:i:0 MD:Z:38 NM:i:0 chr1 123450 123500 + chr5 28374615 28374615 - • Raw FASTQ • Sequence ID, sequence • Quality ID, quality score • Mapped SAM • Map: 0 OK, 4 unmapped, 16 mapped reverse strand • XA (mapper-specific) • MD: mismatch info • NM: number of mismatch • Mapped BED • Chr, start, end, strand http://samtools.sourceforge.net/SAM1.pdf
Data Analysis • Heuristic filtering to identify novel genes for Mendelian disorders Stitziel et al, Genome Biol 2011
Genomic Structural Variation Baker et al, Nat Meth 2012
Structural Variation Detection BreakDancer Chen et al, Nat Meth 2009 Only look at anomalous read pairs
Structural Variation Detection • Crest (Wang et al, Nat Meth 2011) • Use soft-clipped reads, kind of like bidir-blast
Copy Number Variation Detection • Change in read coverage
Representation: VCF Format • http://www.1000genomes.org/node/101
Summary • Whole genome and whole exome sequencing • Solution hybrid selection • Specific locus for rare diseases • Bioinformatics issues: • Read mapping • SNP, indel detection • Heuristic filtering • Structural variation detection