Download

1 / 23
490 likes | 1.24k Views
By: Afif Hossain. Spinal Muscular Atrophy. A Detailed Look at the Mechanism of a Neurodegenerative Disease That Causes Muscle Deterioration. Introduction To SMA. Atrophy- the medical term for wasting or shrinkage, which is what generally happens to muscles when they’re not active

E N D
By: AfifHossain Spinal Muscular Atrophy A Detailed Look at the Mechanism of a Neurodegenerative Disease That Causes Muscle Deterioration
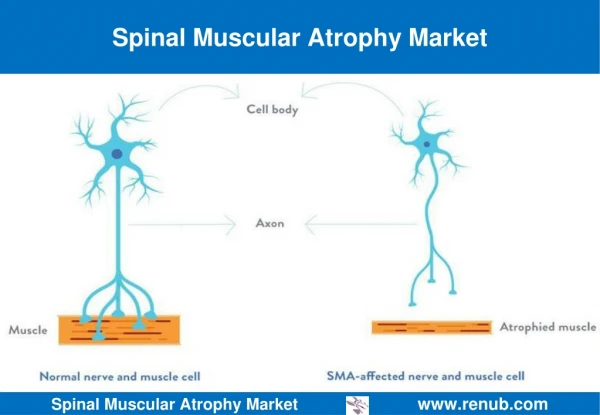
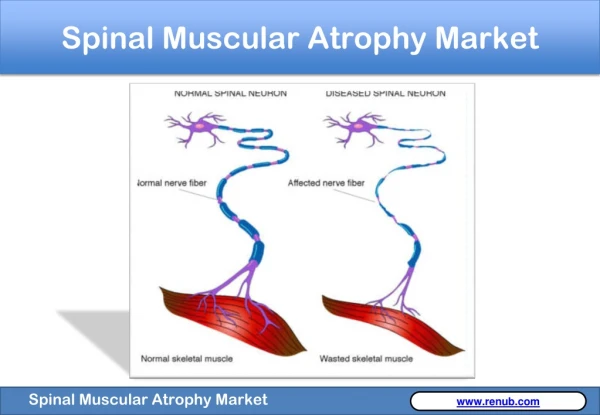
Introduction To SMA • Atrophy- the medical term for wasting or shrinkage, which is what generally happens to muscles when they’re not active • Autosomal recessive disease that affects about 1 in every 6000 individuals • Linked to the survival of motor neuron gene, SMN1 • SMN protein discovered in 1995 • Cause: loss or degeneration of nerve cells in the spine known as motor neurons • Loss of motor neurons is a result of a deficiency in a motor neuron protein called survival of motor neurons, or SMN for short • SMN plays an integral role in normal motor neuron function, and may even directly affect muscle cells themselves1
Introduction to SMA • Mechanism: motor neurons, located in the spinal cord, signal muscle contraction by carrying a signal from the spinal cord, down long, wire-like projections, to muscles • Without ample motor neurons, signal transduction is hindered and muscles cannot function, leading to atrophy • Specifically, mutations on chromosome 5 debilitate the production of SMN protein • Greater levels of SMN protein lessen the severity of the disease, which is loosely related to age (if onset is later, then degeneration of motor neurons will occur later)1 • SMA is entirely genetic
Types of SMA • Type 1 SMA: • Acute and infantile- from birth to 6 months of age • Children often have trouble breathing, swallowing, and sucking and suffer from hypotoniaa • High fatality rates: mean survival is 5.9 months, 95% of cases die by 18 months • Type 2 SMA: • Most common type of SMA, often related with developmental motor delay • Chronic and infantile- onset between 6 and 18 months after birth • Muscles closer to the center of the body (proximal muscles) are usually more affected or earlier (thighs as opposed to the lower legs)1 • Respiratory issues and spinal curvature (scoliosis) also present major problems for future health • Survival into young adulthood or even later can be expected (2 to 30 years)2 a progressive muscle weakness and flaccid or reduced muscle tone
Types of SMA • Type 3 SMA: • Chronic and juvenile- onset of SMA 18 months after birth • Muscular functioning is much more developed and life expectancies are often normal, but respiratory issues and spinal curvature still need to be monitored • Type 4 SMA: • Milder form of SMA that affects adults (normal life expectancies) • There are forms of SMA that are not related to SMN and do not originate from mutations of chromosome 51, but those disorders will not be the focus of my presentation • Mental and emotional development and sensation are entirely normal in SMA
Frequency and Diagnosis • Second most common autosomal-recessive inherited disorder after cystic fibrosis • SMA types I and III each account for about one fourth of cases, whereas SMA type II accounts for one half of all cases by itself • SMA I: Weakness is greater in proximal than distal muscles and may mimic muscle disease (myopathy) and limbs and joints may be deformed at birth • SMA II: Infants cannot get to a sitting position on their own but may retain that position if placed into it; proximal muscle weakness still present • All patients with spinal muscular atrophy retain at least 1 copy of SMN2, which generates only 10% of the amount of full-length SMN protein (compare to SMN1) • Possible therapeutic pathway is to promote SMN2 to function like the missing SMN1 gene2
Social Implications and Funding Bioethical Issues • Disease can be detected in prenatal testing • Sharing genetic information between family members • Family planning- in vitro fertilization • Although SMA cannot be prevented and the symptoms cannot be mitigated, (since it is entirely genetic), testing still allows individuals and families to cope with the disease6 Economic Aspects • Over $60 million available for funding by the Spinal Muscular Atrophy Foundation • $400 for a carrier screen and $260,000 for the lifetime cost of a child with severe disease • Researchers have concluded that 11,000 women would have to be screened to prevent one case of SMA, at a cost of $4.7 million per caseaverted7
Introduction to SMN Tudor domain, 1mhn • The SMN protein plays an important role in the assembly of the spliceosomal small nuclear ribonucleoproteinbcomplexes. • snRNPs are essential to the removal of introns from pre-mRNA, a critical aspect of post-transcriptional modification of RNA • The SMN protein is found in both the nucleus, where it localizes near snRNPs, and the cytoplasm, where the SMN protein plays an important role in the assembly of snRNPs • In order for snRNPs to assemble, they must interact with Smproteinsc • In the Tudor Domain of the SMN protein, the SMN proteins must bind to arginine-glycine (RG) rich C-terminal tails of the Sm proteins in vitro3 bRNA-protein complexes that combine with unmodified pre-mRNA and various other proteins to form a spliceosome, a large RNA-protein molecular complex upon which splicing of pre-mRNA occurs CA protein that belongs to a group of seven core components of splicing small nuclear ribonucleoprotein particles
Figure 1. Structural depiction of the SMN Tudor Domain. One alpha helix is shown between the fourth and fifth beta sheet. The five beta sheets are shown in orange, connected by three loops, shown in blue. The N and C terminals are emphasized with cyan. The antiparallel beta sheets form a beta barrel structure . α1 Figure 1
SMN Tudor Domain and RG Tails • Recent studies show that the Sm proteins, which contain symmetrically dimethylated arginine residues (sDMA), modify sDMAsin vivo • Arginine methylation increases the affinity for the interaction of the SMN Tudor domain with RG repeats, shown by the saturation of the binding site at a much lower RG:proteinratio • The SMN Tudor domain uses the same binding pocket for the interaction with methylated and non-methylated RG tails • Differences in the saturation end points indicate amides (Trp102, Tyr109, Tyr127 and Tyr130) close to sDMA methyl groups, which form the binding pocket • Glycine residues in the methylated RG repeats is postulated to provide conformational stability required for RG-Tudor Domain interactions3
Figure 2. Crystal Structure of the SMN Tudor Domain. (a) Ribbon representation of the SMN Tudor Domain in blue, highlighting the binding pocket in yellow (aromatic side chains include Tryptophan 102 and Tyrosine 109, 127, and 130). Glutamate 134 is shown in green. (b) Close up view of the binding pocket. Figure 2a Figure 2b
SMN Tudor Domain, 1MHN • Modification of sDMA proteins is important because it strongly enhances the affinity of the SMN/Sm interaction and has been implicated in the regulation of U snRNP assembly (similar to methylation, involves Gemin2-7) • Glu134, which when mutated to lysine is linked to type I SMA, is located close to the sDMA binding pocket and abolishes Sm binding in vitro and interferes with snRNP assembly in vivo3 • The point mutation E134K hinders proper recognition of the RG-repeats by the SMN protein and the SMN Tudor domain, which causes SMA I
Introduction to the Gemin6-Gemin7 Heterodimer from the Human SMN Complex • As mentioned before, the SMN complex’s function is assembly of small nuclear ribonucleoproteins (snRNPs), which are major components of spliceosomesd • SnRNPs consists of one U snRNA molecule, a core composed of seven highly conserved Sm proteins, and several snRNP-specific proteins • The major U snRNAs are exported to the cytoplasm where the Sm protein core is assembled on the Sm site of the U snRNA and the 5′-cap is hypermethylated (modification leads to snRNP formation)4 • The SMN complex contains several proteins called Gemins2–7 • Gemin6 and Gemin7 have a two-fold-symmetric heterotetramer symmetry and similar folds, with a five-stranded bent β sheet flanked by α helices, which form a hydrophobic pocket and only allow for highly specific interactions • Since reduced levels of SMN, as a result of deletions or “loss-of-function” mutations in the SMN gene cause SMA, the role of Gemin6/7 is vital to SMN protein production dspecialized RNA and protein subunits that removes introns from a transcribed pre-mRNA segment
Structural View of Gemin6 and Gemin7 Figure 3. Gemin6 in cyan and Gemin7 in orange. Chains highlighted, N and C termini shown in their respective colors. SMN protein binding sites are shown in gray. Having similar folds, with a five-stranded bent β sheet flanked by α helices, the two form a heterodimer connected by β4 of Gemin6 and β5 of Gemin7, resulting in the formation of a continuous 10-stranded β sheet. The longer N-terminal helix of Gemin7 packs tightly into a hydrophobic pocket formed by α1, β2-β4, and α2 of Gemin6, which is flanked by a network of hydrogen bonds formed between α1 side chains in Gemin7 and pocket residues of Gemin6. This corroborates the high specificity to which Gemin6 and Gemin7 interact.4 Figure 3
Similarities Between Gemin6/7 and Sm Proteins • Amino acid sequences in Gemin6 and Gemin7 contain folds that are similar to those observed in the spliceosomalSm proteins • For this reason, Gemin6/7 could serve as an Sm dimer surrogate, binding to individual Sm proteins or to Smsubcomplexes to form a ring-like structure in preparation for snRNA loading • Therefore, Gemin6 and Gemin7 have been shown to interact in vitro with the human Sm proteins, binding and organizing Sm proteins in preparation for snRNPassembly. • This correlates to both the proposed theory that human Sm proteins form heptameric rings when assembled on the Sm sites of U snRNAs and the function of symmetrically dimethylated arginine residues in the RG-rich tails to recognize Smproteins • Gemin6/7 facillitatesSm/SMN protein interactions4
Sm Protein and Gemin6/7 Interactions (b) Specific residues of interaction. In SmB/B’, Asn39 hydrogen bonds with Gly74 and Asp35, linking the C-terminal segments of strands β2 and β4 with the N terminus of β3. In Gemin6, Asn43, Gly62, and Asp38 form an identical hydrogen bonding network. Similarly, SmD3 residues Asp37, Asn40, and Tyr62 and Gemin7 residues Asp96, Asn101, and Tyr103 form the same hydrogen bonding patterns4. Figure 4b Figure 4a Figure 4. Mechanism that Sm proteins interact with Gemin 6/7. (a) There are two binding surfaces that Gemin6/7 could bind to Sm proteins: The β5 surface of Gemin6 and the β4 surface of Gemin7 are both exposed and available for interaction with Sm proteins in the Gemin6/7 complex. Sm Proteins represented with arrows.
Human DcpS (3BL9) and Its Mechanism • Although SMA is determined by a mutation or deletion in both copies of the SMN1 gene, the severity of SMA is modified by the second gene, SMN2, that produces an mRNA that is incorrectly spliced with the deletion of the last exon • When SMN2 has a point mutation, it can only produce 10% of the SMN protein necessary for normal muscular functioning • DcpSmodulates gene expression at the level of mRNA turnover and pre-mRNA splicing • DcpSfunctions in the last step of mRNA decay to hydrolyze the mRNA cap structure (m7GpppN) following 3′to 5′ exonucleolytic decay and 5’ end decapping, which causes mRNA decay (dephosphorylates GDP to GMP in the residual cap structure, reducing the energy necessary for the mRNA to undergo translation)5 • DcpS continues to decap mRNAs that lack a stop codon, causing SMN2 protein levels to decrease
In order for inhibition to be effective, both the open and closed active sites must be inhibited. Since D157493 can only bind to the closed active site, a point mutation from Histadine to Asparanine inhibits the open active site. (d) (a) (c,d) The open active site is conformationally “open” because it is surrounded by α helices, whereas the closed active site is conformationally “closed”, because it is tangled in β sheets5. Figure 5. (a) Active site His277Asn mutation in complex with m7GpppG substrate and inhibitor (c) (b) m7GpppG base occupies a narrow pocket staking between Leu206 and Trp175, and also forms hydrogen bonds to the Glu185 side chain and the carbonyl oxygen of Pro204 (b)
Possible Treatments: D157493 (C5-quinazolines) • Although the exact role and mechanism of DcpS in relation to the SMN protein is not clearly understood, there is a general consensus that DcpS is the therapeutic target of SMA because its role in inhibiting the production of the SMN protein by closing the SMN protein’s substrate-bound complex and opening its product-bound complex • Therefore, a group of drugs called C5-quinazolines, commonly referred to as D157493, are currently being tested for their therapeutic value in treating SMA • C5-quinazolines are experimental drug compounds that are in vitropotent DcpS inhibitors, causing increases in SMN mRNA levels in SMA and corresponding increases in SMN protein production (by inhibiting DcpS at its active sites) • Other drugs, like histone deacetylase inhibitors, including valproic acid have also been shown to increase transcription of the SMN2 gene and thus increase levels of the SMN protein5
References Video Clip: Fight Spinal Muscular Atrophy.FightSMA. http://www.youtube.com/watch?v=aZUVFRAyl_I • Muscular Dystrophy Association. “Facts About Spinal Muscular Atrophy (SMA).” MDA Publications. 2010. • Tsao, Bryan and Armon, Carmel. “Spinal Muscular Atrophy.” eMedicine. 14 Jan 2009. • Sprangers, Remco, Groves, Matthew R., Sinning, Irmgardand Sattler, Michael. “High-resolution X-ray and NMR Structures of the SMN Tudor Domain: Conformational Variation in the Binding Site for Symmetrically Dimethylated Arginine Residues.” Science Direct. March, 2003.
References Cont’ • Ma, Yingli, Dostie, Josée, Dreyfuss, Gideon and Van Duyne, Gregory D. “The Gemin6-Gemin7 Heterodimer from the Survival of Motor Neurons Complex Has an Sm Protein-like Structure.” Science Direct. July 2005. • Singh, Jasbir, Salcius, Michael, Liu, Shin-Wu, and Staker, Bart L. “DcpS as a Therapeutic Target for Spinal Muscular Atrophy.” National Institute of Health Public Access. November 2008. • Norrgard, Karen. “Medical Ethics: Genetic Testing and Spinal Muscular Atrophy.” Scitable. Web. 2008. • Preidt, Robert. “Screening for Spinal Muscular Atrophy Not Cost-Effective: Study.” MedicineNet.com. Web. 2 Februrary, 2010.