Download

1 / 36
390 likes | 701 Views
Spinal Muscular Atrophy. the number one genetic killer of infants and toddlers. Leena Shah Human Anatomy April 21, 2010. Samir died at 7 ½ months. Hailey Mae died at the age of 2. Owen died at 19 weeks and 6 days. Baylee died at 15 months. Spinal Muscular Atrophy (SMA).

E N D
Spinal Muscular Atrophy the number one genetic killer of infants and toddlers Leena Shah Human Anatomy April 21, 2010
Samir died at 7 ½ months Hailey Mae died at the age of 2 Owen died at 19 weeks and 6 days Baylee died at 15 months
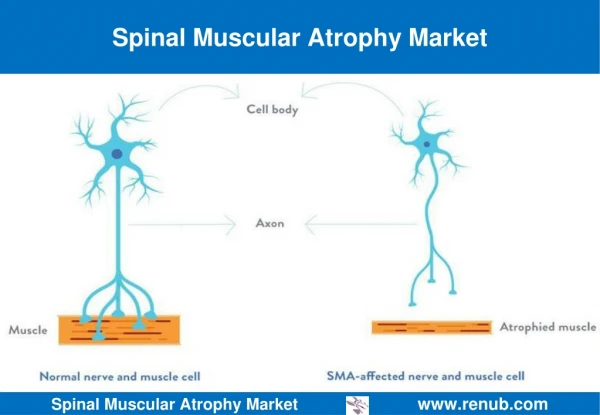
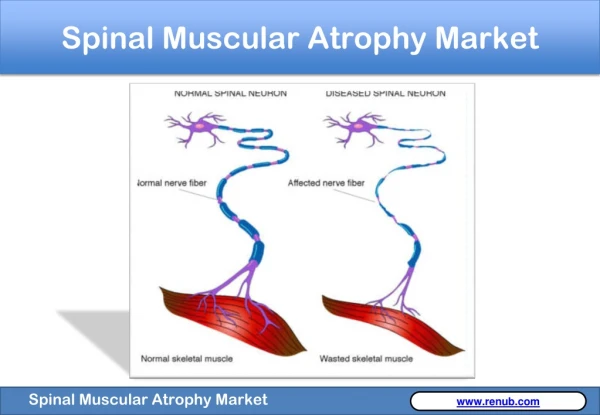
Spinal Muscular Atrophy (SMA) • A genetic disease in which loss of nerve cells in the spinal cord called motor neurons affects the part of the nervous system that controls voluntary muscle movement
Mouse Model of Spinal Muscular Atrophy • Motor neuron axons (green) • neuromuscular junctions (red) Both mice are 11 days old. The mouse on the left does not have SMA. The SMA mouse on the right is much smaller due to effects of the disease, and it is unable to move normally or fully support its weight
Symptoms and Diagnosis • Weakened muscles • Respiratory problems • Scoliosis • Difficulty walking and sitting • Diagnosis of SMA is made based upon physical symptoms5 • poor muscle tone in the limbs and trunk • feeble movements of the arms and legs • swallowing difficulties • a weak sucking reflex • impaired breathing
Four Types of SMA • SMA Type I • also called Werdnig-Hoffman disease • is the most severe form (lifespan 2-3 years) • symptoms are present before the age of six months and these children never acquire the power, the strength, and the endurance to sit independently, to crawl, walk, or breath properly • SMA I affects 60% of all SMA patients • SMA Type II • less severe than type 1 • comes to medical attention before the age of 18 months • Respiratory muscles and feeding problems not as severe as in Type 1 • Can sit, but still cannot walk properly • Can live into adulthood • SMA Type III • Known as Kugelberg-Welander disease or Juvenile Spinal Muscular Atrophy • Symptoms appear between 18 months and early adulthood • Difficulty walking, muscles weakness, prone to respiratory infections • Normal life expectancy • SMA Type IV • Adult form of SMA – less common • Affects walking • Symptoms emerge after age 35 3
The Survival of Motor Neuron (SMN) Gene • Spinal Muscular Atrophy is caused by mutations in the SMN gene • SMN gene located on chromosome 5 • Autosomal recessive gene • Gene codes for the SMN protein 3
Survival of Motor Neuron Protein • Most SMN proteins are localized in the cytoplasm • They play a crucial role in the assembly of spliceosomal uridine-rich small nuclear ribonucleoprotein (U snRNP) complexes • snRNPs are essential to the splicing of introns from pre-mRNA during the post-transcriptional modification of RNA • snRNPs are made up of small nuclear RNA (snRNA) and a group of seven proteins known as Sm ribonucleoproteins that make up the extremely stable Sm core of the snRNP • How do SMN proteins assemble these U snRNP complexes? 4
The Formation of snRNPs • snRNA interacts with the SMN protein to form a SMN complex • The SMN complex functions as an assembly machine for snRNPs • It binds with Sm proteins and organizes them so that they can bind with snRNA to form snRNPs • The Sm core of snRNPs consists of seven proteins: B, D1, D2, D3, E, F and G • SMN protein acts as a chaperone to ensure that the Sm core assembles onto the correct snRNA • The Sm proteins form a seven-member ring core structure that encircles the snRNA 4
Central Tudor Domain of the Human SMN protein N • SMN proteins assemble U snRNPs via binding to Sm core proteins • The Human SMN protein contains a central Tudor domain that facilitates this SMN–Sm protein interaction7 Loop 2 β1 β2 β5 C β3 β4 Figure 1.Tudor Domain of the Human SMN Protein (residues 83-173). The structure forms a bent antiparallel β-sheet. Five β strands, β1, β2, β3, β4, and β5, form a barrel-like fold (labeled in blue). The strands are connected with short turns labeled in orange. Loop 1 is in between β1-β2, Loop 2 is in between β2-β3, and Loop 3 is in between β3-β4. The N-terminus and C-terminus are also labeled in orange. Loop 3 Loop 1
Interaction with Sm Proteins • SMN binds to the Arg and Gly-rich C-terminal tails of the Sm D1 and D3 proteins • When the Tudor domain has mutation on Glutamic Acid residue 134 [E134K], (a point mutation that causes SMA), SMN binds to neither • Glutamic Acid (E) is changed to Lysine (K) • Therefore it can be concluded that the Tudor domain binds to the C-terminal tails of Sm D1 and D3 7
The E134K Mutation N C Loop 2 β5 β1 β2 E134K Figure 2. The Tudor Domain of the Human SMN Protein is shown. Individual β strands are labeled in cyan. Turns between βstrands (Loops 1, 2, and 3) are labeled in orange along with the N-terminus and C-terminus. In blue is the Glutamic Acid residue 134. During an E134K mutation, the Glutamic Acid residue (E) would be changed to a Lysine residue (K). β3 β4 Loop 3 Loop 1
How does the E134K mutation impair Sm binding? • E134K mutant does not disrupt the three-dimensional structure of the Tudor domain • several negatively charged amino acids are located in loop 1 (Glu 104, Asp 105), β4 (Glu 134) and the helical turn connecting β4 and β5 (Asp 140) • These residues are conserved in all SMN homologs causing the structure to exhibit an overall negatively charged surface 7
Negatively Charged Amino Acids β5 Asp 140 β4 3b • Figure 3. • The Tudor Domain of the Human SMN Protein with negatively charged amino acids highlighted. Loop 1 has Glutamic Acid residue 104 in orange and Aspartic Acid residue 105 in cyan. Glutamic Acid residue 134 is in blue on β4. The Aspartic Acid residue 140 is located between β4-β5 in yellow. • Structure of the Tudor Domain like 3a. Figure shows all the side chains of the negatively charged amino acids highlighted in 3a. Glu 134 Loop 1 Asp 105 Glu 104 3a
Mutation Disrupts Charge Distribution • the E134K mutation changes the local charge distribution at the Sm binding site • This affects electrostatic interactions with the positively charged C-terminal Sm tails7
Asp 140 Glu 134 Asp 105 Glu 104 Figure 4. Surface structure of the Tudor Domain of the Human SMN Protein. On the left, negatively charged residues Glu 134 (in blue), Glu 104 (in orange), Asp 105 (in cyan), and Asp 140 (in yellow) are shown. On the right, the charge distribution of the same Tudor domain is shown, with the negatively charged surfaces in red. Therefore if Glu 134 were to be mutated, it would affect the charge distribution and electrostatic interactions with other positively charged proteins.
Distal Spinal Muscular AtrophyType V • Other forms of spinal muscular atrophy are caused by mutation of other genes • One form is the disease Distal SMA, type V • caused by missense mutations on the enzyme glycyl-tRNA synthetase (GlyRS) • This progressive disorder affects nerve cells in the spinal cord and weakens muscles in the hands and feet • Also known as Distal SMA with upper limb predominance 2
Symptoms of Distal SMA Type V • Usually begin during adolescence • Initial symptom – cramps in the hand induced by exposure to cold temperatures • Weakness and wasting away (atrophy) of hand muscles, especially between index finger and thumb • Foot abnormalities • High arch (pes cavus) • affected individuals eventually develop problems with walking (gait disturbance) • People have normal life expectancies 2
Glycyl-tRNA Synthetase (GlyRS) • GlyRS is an enzyme in humans coded by GARS gene • autosomal dominant pattern • one copy of the altered gene in each cell is sufficient to cause the disorder • GlyRS is a type of aminoacyl-tRNA synthetase (ARS) 1
Aminoacyl-tRNA Synthetase (ARS) • ARS are enzymes which couple amino acids with their cognate tRNAs to form an aminoacyl-tRNA • This allows the translation from genetic code to amino acid code during translation
GlyRS with mutation S581L • Serine residue 581 missense mutation is 50 amino acids from the active site • Serine (S) is replaced with Leucine (L) at residue 581 • Yet gives reduced aminoacylation activity • Interesting because patients carrying this mutation develop disease younger age than for other GlyRS mutations • The S581L mutation is located on an alpha-helix in the anticodon-binding domain and induces a local conformation change • Overall structure between mutant and wildtype similar but residues 567-575 of the anticodon-binding domain have shift position 1
Residues 567-575 Glycyl-tRNA Synthetase GlyRS with mutation S581L Figure 5. Molecular Structure of wildtype and S581L GlyRS mutant. On the left, the wildtype structure is present with residues 567-575 highlighted in white. On the right, the S581L mutant structure is present with, again, residues 567-575 in white. A small shift in position resulting in the localized conformation of the protein is seen. The area where the conformation occurs is known as the anticodon-binding domain of the GlyRS protein.
Residues 567-575 Glycyl-tRNA Synthetase GlyRS with mutation S581L Figure 6. Surface Structure of wildtype and S581L GlyRS mutant. On the left, the wildtype surface structure is present with residues 567-575 highlighted in white. On the right, the S581L mutant surface structure is present with, again, residues 567-575 in white. A more prominent shift in position resulting in the localized conformation of the protein is seen. The conformation occurs in the anticodon-binding domain of the GlyRS protein.
Reduced GlyRS Activity • The S581L mutation results in the loss of contact with the following atoms: • L580, A583 and T585 • Additional contacts are made with following atoms: • V564, V577, and H591 • The mutation therefore causes a small shift in the adjoining beta-turn of residues 567– 575 which is a position that interacts with the tRNA anticodon • The shifted residues are displaced away from the tRNA in S581L-GlyRS compared to wildtype • This shift can affect glycine binding to the tRNA reduced enzyme activity of GlyRS = reduced aminoacylation 1
Affected Residues V564 H591 L580 S T585 V577 A583 L S581L Figure 7. A close up of the wildtype GlyRS Protein is shown on the left. The Serine residue 581, (where the S581L mutation would occur), is highlighted in green along with its side chain. In cyan are residues L580, A583, and T585 with their side chains. Contact with these residues is lost during an S581L mutation. In yellow are residues V564, V577, and H591 with their side chains. New contacts with these residues are made during an S581L mutation. Side chains are coded by their elemental makeup (red for oxygen, gray for carbon, blue for nitrogen). At the top right the structure of the GlyRS S581L mutant is shown. The picture is identical to the wildtype with the exception of the Serine (S) 581 residue mutated to Lysine (L). The difference in the side chains can be see on the bottom right as Lysine has no oxygen and is a nonpolar residue as opposed to Serine, which is polar.
Treatment • There is no treatment for the progressive muscle weakness caused by the disease • Respiratory complications are common • Physical therapy is important to prevent contractions of muscles and tendons and abnormal curvature of the spine (scoliosis) 3
A Therapeutic Target: Scavenger Decapping Enzyme (DcpS) • SMA is caused by mutation in SMN1 gene • The severity of SMA involves a second gene, SMN2 • DcpS is a potential binder to the SMN2 promoter • DcpS functions in the last step of mRNA decay to hydrolyze the cap structure following 3’ to 5’ exonucleolytic decay • DcpS can be used to modulate gene expression of SMN2 gene 8
DcpS Basics • Before translation, mRNA Processing • One of the alterations is the addition of a 5’ cap • Capping provides stability to mRNA as well as a point of attachment to the ribosome during translation • DcpS binds to the SMN2 transcript • Decapping enzyme DcpS degrades RNA by hydrolyzing the cap structure and thereby splicing the last exon (7), the last coding exon in the SMN2 transcript • This results in the translation of an incorrectly spliced SMN2 mRNA and the formation of less SMN2 proteins than normal 8
DcpS bounded to Inhibitor Drug D156844 • DcpS has two active sites: a closed one and an open one • D156844 is C5-substituted quinazoline • It inhibits the closed active site of DcpS while a point mutation His277Asn inhibits the open active site • By inhibiting DcpS, the SMN2 mRNA is not decapped and exon 7 is not spliced, resulting in increased SMN2 mRNA levels • This causes increased SMN2 levels and a decrease in the severity of SMA disease 8
DcpS bound to Inhibiter D156844 D156844 His 277 Figure 8. Molecular Structure of Scavenger Decapping Enzyme (DcpS) bound with inhibitor D156844. Also, highlighted in blue, is residue 277 – the location of where the point mutation of Histidine to Asparanine would occur.
DcpS Inhibited by D156844 at the Closed Active Site D156844 Figure 9. A close up of the Molecular Structure of DcpS. Bound to the molecule is the inhibiter D156844. The position of the inhibitor is on an active site that is “closed” because of its tight conformation.
DcpS Open Active Site His277 Figure 10. A close up of the molecular structure of DcpS with the potential location of mutation His277Asn highlighted. The mutation would occur in the “open” active site of the DcpS. (open because of loose conformation).
Social Implications of SMA • Mutation causing SMA can be detected via prenatal testing5 • Genetic counselor – informs of choices, risks, benefits • Prepare for an affected baby with early intervention breathing and physical therapies for the newborn • SMA affects every 1 in 6000 babies5 • In Vitro Fertilization (IVF) • only those embryos without SMA are transferred back to the mother's uterus • Chorionic Villus Sampling (CVS) • a sample of the placenta is removed via catheter at 10-12 weeks of gestation • Test during ongoing pregnancy and then faced with the decision of whether or not to terminate the pregnancy • Early vs. Late Diagnosis • Newborn screening can allow parents to start with intervention strategies right away before waiting for symptoms to make a diagnosis • Is there enough benefits from early testing to counteract the cost of testing every newborn? • No effective treatments to delay or prevent symptoms found yet5 • In 2009 costs were estimated to be $400 for a carrier screen and $260,000 for the lifetime cost of a child with severe SMA6 • Researchers concluded that 11,000 women would have to be screened to prevent one case of SMA – a cost of $4.7 million per every averted SMA case6
References • Cader, Muhammed Z., et al. "Crystal Structure of Human Wildtype and S581L-Mutant Glycyl-tRNA Synthetase, an Enzyme Underlying Distal Spinal Muscular Atrophy." FEBS letters 581.16 (2007): 2959-64. Print. • "Distal Hereditary Motor Neuropathy, Type V - Genetics Home Reference." Genetics Home Reference - Your Guide to Understanding Genetic Conditions. U.S. National Library of Medicine, Aug. 2009. Web. 21 Apr. 2010. <http://ghr.nlm.nih.gov/condition=distalhereditarymotorneuropathytypev>. • "Frequently Asked Questions." SMA Foundation | Spinal Muscular Atrophy. Spinal Muscular Atrophy Foundation. Web. 21 Apr. 2010. <http://www.smafoundation.org>. • McDowall, Jennifer. "Sm Ribonucleoproteins." European Bioinformatics Institute | Homepage | EBI. Protein Data Bank. Web. 21 Apr. 2010. <http://www.ebi.ac.uk/interpro/potm/2005_5/Page1.htm>. • Norrgard, Karen. “Medical Ethics: Genetic Testing and Spinal Muscular Atrophy.” Scitable. Web. 2008. • Preidt, Robert. "Screening for Spinal Muscular Atrophy Not Cost-Effective: Study: MedlinePlus." National Library of Medicine - National Institutes of Health. 5 Feb. 2010. Web. 21 Apr. 2010. <http://www.nlm.nih.gov/medlineplus/news/fullstory_94965.html>. • Selenko, Philipp, et al. "SMN Tudor Domain Structure and its Interaction with the Sm Proteins." Nat Struct Mol Biol 8.1 (2001): 27-31. Print. • Singh, Jasbir, et al. "DcpS as a Therapeutic Target for Spinal Muscular Atrophy." ACS Chemical Biology 3.11 (2008): 711-22. Print.