Download
1 / 1
30 likes | 265 Views
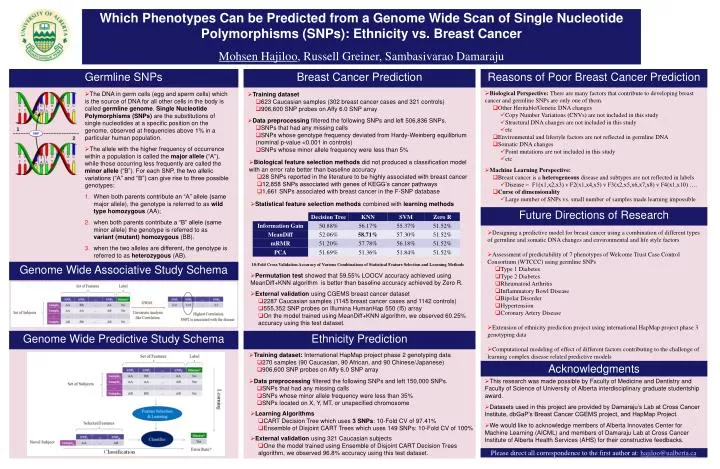
Which Phenotypes Can be Predicted from a Genome Wide Scan of Single Nucleotide Polymorphisms (SNPs): Ethnicity vs. Breast Cancer Mohsen Hajiloo , Russell Greiner, Sambasivarao Damaraju. Germline SNPs. Breast Cancer Prediction. Reasons of Poor Breast Cancer Prediction.

E N D
Which Phenotypes Can be Predicted from a Genome Wide Scan of Single Nucleotide Polymorphisms (SNPs): Ethnicity vs. Breast Cancer Mohsen Hajiloo, Russell Greiner, Sambasivarao Damaraju Germline SNPs Breast Cancer Prediction Reasons of Poor Breast Cancer Prediction 10-Fold Cross Validation Accuracy of Various Combinations of Statistical Feature Selection and Learning Methods • The DNA in germ cells (egg and sperm cells) which is the source of DNA for all other cells in the body is called germline genome. Single Nucleotide Polymorphisms (SNPs) are the substitutions of single nucleotides at a specific position on the genome, observed at frequencies above 1% in a particular human population. • The allele with the higher frequency of occurrence within a population is called the major allele (“A”), while those occurring less frequently are called the minor allele (“B”). For each SNP, the two allelic variations (“A” and “B”) can give rise to three possible genotypes: • When both parents contribute an “A” allele (same major allele), the genotype is referred to as wild type homozygous (AA); • when both parents contribute a “B” allele (same minor allele) the genotype is referred to as variant (mutant) homozygous (BB). • when the two alleles are different, the genotype is referred to as heterozygous (AB). • Biological Perspective: There are many factors that contribute to developing breast cancer and germline SNPs are only one of them. • Other Heritable/Genetic DNA changes • Copy Number Variations (CNVs) are not included in this study • Structural DNA changes are not included in this study • etc • Environmental and lifestyle factors are not reflected in germline DNA • Somatic DNA changes • Point mutations are not included in this study • etc • Training dataset • 623 Caucasian samples (302 breast cancer cases and 321 controls) • 906,600 SNP probes on Affy 6.0 SNP array • Data preprocessing filtered the following SNPs and left 506,836 SNPs. • SNPs that had any missing calls • SNPs whose genotype frequency deviated from Hardy-Weinberg equilibrium (nominal p-value <0.001 in controls) • SNPs whose minor allele frequency were less than 5% • Biological feature selection methods did not produced a classification model with an error rate better than baseline accuracy • 28 SNPs reported in the literature to be highly associated with breast cancer • 12,858 SNPs associated with genes of KEGG’s cancer pathways • 1,661 SNPs associated with breast cancer in the F-SNP database • Machine Learning Perspective: • Breast cancer is a heterogeneous disease and subtypes are not reflected in labels • Disease = F1(x1,x2,x3) v F2(x1,x4,x5) v F3(x2,x5,x6,x7,x8) v F4(x1,x10) …. • Curse of dimensionality • Large number of SNPs vs. small number of samples made learning impossible • Statistical feature selection methods combined with learning methods Future Directions of Research • Designing a predictive model for breast cancer using a combination of different types of germline and somatic DNA changes and environmental and life style factors • Assessment of predictability of 7 phenotypes of Welcome Trust Case Control Consortium (WTCCC) using germline SNPs • Type 1 Diabetes • Type 2 Diabetes • Rheumatoid Arthritis • Inflammatory Bowl Disease • Bipolar Disorder • Hypertension • Coronary Artery Disease • Extension of ethnicity prediction project using international HapMap project phase 3 genotyping data • Computational modeling of effect of different factors contributing to the challenge of learning complex disease related predictive models Genome Wide Associative Study Schema • Permutation test showed that 59.55% LOOCV accuracy achieved using MeanDiff+KNN algorithm is better than baseline accuracy achieved by Zero R. • External validation using CGEMS breast cancer dataset • 2287 Caucasian samples (1145 breast cancer cases and 1142 controls) • 555,352 SNP probes on Illumina HumanHap 550 (I5) array • On the model trained using MeanDiff+KNN algorithm, we observed 60.25% accuracy using this test dataset. Genome Wide Predictive Study Schema Ethnicity Prediction • Training dataset: International HapMap project phase 2 genotyping data • 270 samples (90 Caucasian, 90 African, and 90 Chinese/Japanese) • 906,600 SNP probes on Affy 6.0 SNP array Acknowledgments • Data preprocessing filtered the following SNPs and left 150,000 SNPs. • SNPs that had any missing calls • SNPs whose minor allele frequency were less than 35% • SNPs located on X, Y, MT, or unspecified chromosome • This research was made possible by Faculty of Medicine and Dentistry and Faculty of Science of University of Alberta interdisciplinary graduate studentship award. • Datasets used in this project are provided by Damaraju’s Lab at Cross Cancer Institute, dbGaP’s Breast Cancer CGEMS project, and HapMap Project. • We would like to acknowledge members of Alberta Innovates Center for Machine Learning (AICML) and members of Damaraju Lab at Cross Cancer Institute of Alberta Health Services (AHS) for their constructive feedbacks. • Learning Algorithms • CART Decision Tree which uses 3 SNPs: 10-Fold CV of 97.41% • Ensemble of Disjoint CART Trees which uses 149 SNPs: 10-Fold CV of 100% • External validation using 321 Caucasian subjects • One the model trained using Ensemble of Disjoint CART Decision Trees algorithm, we observed 96.8% accuracy using this test dataset. Please direct all correspondence to the first author at: hajiloo@ualberta.ca