Download

1 / 1
10 likes | 93 Views
Explore the use of Caenorhabditis elegans as an infection model to study genes shared between pathogens and hosts. Investigate unusual bacterial-eukaryotic homologs via phylogenetic analysis of C. elegans proteins. Results reveal conserved paralogous proteins and rule out recent horizontal gene transfer. Benefit from Genomics and Bioinformatics for Pathogenomics.

E N D
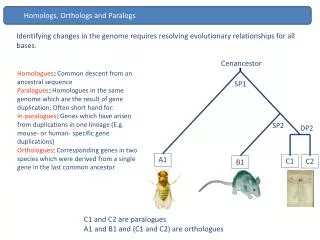
Caenorhabditis species as an infection model for the investigation of genes conserved betweenpathogens and their hosts Nancy L. Price1, Fiona S.L. Brinkman1,2, Steve J.M. Jones3, Hans Greberg3, B. Brett Finlay2, and Ann M. Rose1 1Dept. of Medical Genetics, 2Dept. of Microbiology and Immunology, University of British Columbia V6T 1Z3, Canada, 3Genome Sequence Centre, BC Cancer Agency, Vancouver, British Columbia, V5Z 4E6 Canada Identifying Unusual Bacterial- Eukaryotic Homologs: Focus on C. elegans C. elegans genome analysis: Using BAE-watch, coupled with further phylogenetic analysis, we analyzed all C. elegans proteins that are most similar to bacterial proteins. Phylogenetic analysis, coupled with analysis of proteins of organellar function, indicated that these unusual bacterial-eukaryotic homologs are not the result of recent horizontal gene transfer between ancestors of C. elegans and a given bacterial species. (Note that some eukaryotic organelle genes that have migrated to the nucleus will share highest similarity with bacterial proteins, due to the bacterial ancestry of organelles) A summary of these results is presented: Total number of C. elegans proteins analyzed: 17123 C. elegans proteins with a top BLAST hit to a bacterial protein: 126 C. elegans proteins with a top BLAST hit to a bacterial protein, after proteins of the same family, Rhabditidae, are ignored: 127 C. elegans proteins with a top BLAST hit to a bacterial protein, after proteins of the same phylum, Nematoda, are ignored: 128 C. elegans proteins with a top BLAST hit to a bacterial protein, after proteins of the same kingdom, Metazoa, are ignored: 458 Number of the above 458 proteins with approximately >45% identity to the bacterial protein (MaxRatio>40)*: 44 Number of the above 458 proteins with notably more similarity to bacterial proteins over eukaryotic proteins, as confirmed in phylogenetic analysis, after removal of proteins of probable organelle origin: 2 (Accession: P34275 and O01502in tree below) P34275 and O01502 share a relationship with the same bacterial protein (see tree). However these proteins, all possible acyl-CoA dehydrogenases (by similarity), are members of a large group of highly conserved parologous proteins and the level of similarity between the bacterial and eukaryotic proteins is not consistent with horizontal gene transfer. *Approximately 45% identity between proteins reflects a BAE-watch MaxRatio score of 40, where the MaxRatio = The ratio of the C. elegans BLAST score against itself, verses the C. elegans BLAST score against its top BLAST hit. The higher the MaxRatio, the more similar the proteins. Q9K6D0 Bacillus halodurans B MMGC P12007 Rattus norvegicus (Rat) E IVD Q9JHI5 Mus musculus (Mouse) E IVD P26440 Homo sapiens (Human) E IVD Q9I391 Pseudomonas aeruginosa B PA1631 Q21243 Caenorhabditis elegans E K05F1.3 Q9VSA3 Drosophila melanogaster (Fruit fly) E CG12262 P11310 Homo sapiens (Human) E ACADM P45952 Mus musculus (Mouse) E ACADM P08503 Rattus norvegicus (Rat) E ACADM O28976 Archaeoglobus fulgidus A AF1293 O29413 Archaeoglobus fulgidus A AF0845 O28039 Archaeoglobus fulgidus A AF2244 O29236 Archaeoglobus fulgidus A AF1026 Q9HQF0 Halobacterium sp. (strain NRC-1) A ACD3 OR VNG1191G Q9HS75 Halobacterium sp. (strain NRC-1) A ACD1 OR VNG0371G Q9L079 Streptomyces coelicolor B ACDB P79274 Sus scrofa (Pig) E ACADL P28330 Homo sapiens (Human) E ACADL P15650 Rattus norvegicus (Rat) E ACADL P51174 Mus musculus (Mouse) E ACADL Q9HVY0 Pseudomonas aeruginosa B PA4435 Q9X7Y2 Streptomyces coelicolor B SC6A5.36 Q9I3H8 Pseudomonas aeruginosa B PA1535 O86319 Mycobacterium tuberculosis B FADE13 Q9HZV8 Pseudomonas aeruginosa B PA2889 Letters A, B, and E after each organism name specifies whether the organism is Archaea, Bacteria or Eukarya P34275 Caenorhabditis elegans E C02D5.1 O01502 Caenorhabditis elegans E C37A2.3 Q9I0T2 Pseudomonas aeruginosa B PA2552 Q9HRI6 Halobacterium sp. (strain NRC-1) A ACD4 OR VNG0679G Q9VDT1 Drosophila melanogaster (Fruit fly) E CG4703 P15651 Rattus norvegicus (Rat) E ACADS P16219 Homo sapiens (Human) E ACADS O34421 Bacillus subtilis B YNGJ Q9R9I6 Bacillus subtilis B YNGJ 0.1 Orthologous? Brackets mark proposed orthologous sequences (sequences that diverged due to speciation, rather than gene duplication or horizontal transfer) www.pathogenomics.bc.ca Introduction Genomics and bioinformatics provide powerful new tools for the study of microbial pathogenicity, hence the development of a new field, Pathogenomics. Our Pathogenomics project utilizes a combination of informatics, evolutionary biology, microbiology and eukaryotic genetics to identify pathogen genes which are more similar to host genes than expected, and likely to interact with, or mimic, their host’s gene functions. Currently, our project has been divided into two complementary fields of phylogenetic and functional analysis. Within the phylogenetic analysis, we have developed software which aids identification of horizontally acquired sequences in hope that this approach will enabled us to not only identify new potential virulence factors, but also gain insight into the frequency of horizontal gene transfer within the bacteria, and between the three domains of life of Bacteria, Eukarya, and Archaea. Candidate virulence factors identified by our informatics approach are being targeted for further functional study using a Caenorhabditis model for infection. The utilization of Caenorhabditis as a model organism offers numerous advantages for functional genetic analysis including its small size, ease of maintenance, transparent morphology, rapid generation time, completely sequenced genome, and the large availability of well-developed genetic and molecular tools. In addition, recent published literature has demonstrated that C. elegans can be successfully infected with Pseudomonas aeruginosa, Bacillus megaterium, and Salmonella typhimurium, thus demonstrating C.elegans as a suitable host model for functional analysis of virulent genes during the infection process. Caenorhabditis as a model for infection Rationale: Previous literature has demonstrated successful infection in C. elegans using pathogens such as Pseudomonasaeruginosa, Bacillus megaterium, and Salmonella typhimurium, therefore we rationalized that it would be feasible to establish similar infection models using C. elegans and enteric bacteria such as Yersinia enterocolitica and Listeria monocytogenes. Infection Assay Attempts: Initial infection assays between C. elegans and Y. enterocolitica failed to establish a successful infection model and we investigated several possible factors that would contribute to lack of bacterial infection within the C. elegans host model. These factors included choice of liquid and solid media, pH, salt content, and incubation temperature. Utilization of Thermotolerant Caenorhabditis species Temperature Incubation Dilemma: The optimal growth temperature for C. elegans is 20oC and the maximum temperature that C. elegans remains viable and fertile is 25oC. Temperatures exceeding 25oC result in worm infertility and death. However, virulent gene expression in enteric pathogens is regulated by temperature, with the optimal temperature being 37oC. For example, in Yersinia species, the virulent genes yadA and psaA, as well as the yop operons, are up-regulated at 37oC and down-regulated at temperatures below 26oC. We theorized that a possible explanation of unsuccessful infection of C. elegans with a pathogenic enteric bacterium could be due to the lack of virulent gene expression during room temperature incubation. To circumvent this problem, a compromise between the optimal bacterial temperature and the optimal nematode temperature was suggested and we proposed the utilization of a thermotolerant worm. C. briggsae as a Thermotolerant Host:The search for Caenorhabditis mutants that are capable of remaining viable and fertile at higher temperatures than 25oC, resulted in the acquisition of two Caenorhabditis species namely, a C. elegans daf-2 mutant and the wildtype C. briggsae, var. Gujarati, G16. We performed thermotolerant testing on both candidate Caenorhabditis species to evaluate the maximum temperature each organism could survive and remain fertile. From our analysis, we determined that C. briggsae exhibited the highest thermotolerance, remaining viable and fertile at 30oC after 72 h. Genetic Analysis of the Pathogenecity Process: Currently we are utilizing C. briggsae G16 as the host model of choice in our infection assays with Y. enterocolitica and L. monocytogenes at 30oC with the goal of successfully establishing an infection model. Once established, functional analysis of the putative gene products that are conserved within C. elegans and bacteria will be performed to elucidate possible virulent factors that are involve during the infection process. Identifying Unusual Bacterial- Eukaryotic Homologs Rationale: Pathogen proteins have been identified that manipulate host cells by interacting with, or mimicking, host proteins. We wondered whether we could identify selected novel virulence factors by identifying bacterial pathogen genes more similar to host genes than you would expect based on phylogeny. A web-based tool we developed investigates this, producing a database of such proteins. It is also useful for identifying cross-domain lateral gene transfer events between the three domains of life of Bacteria, Archaea and Eukarya, hence we named this database “BAE-watch”. This tool was used to aid identification of any possible cases of cross-domain horizontal gene transfer between all complete bacterial and eukaryotic genomes, including the C. elegans genome. Description of the BAE-watch database: Proteins in a given pathogen genome that are more similar to eukaryote proteins than other proteins (and vice versa) are identified through BLAST analysis, followed by use of a “StepRatio” scoring system we developed (to screen out of the analysis most proteins that are highly conserved in all organisms, that BLAST may list as most similar to a protein from another Domain by chance). Various taxonomic levels of organisms are filtered from the BLAST results to aid identification of putative lateral transfers that occurred before or after species, genus, family etc… divergence. This database includes an analysis of the C. elegans genome (see next poster section). Analysis of complete bacterial genomes: A comprehensive analysis of all complete bacterial genomes for eukaryotic homologs, using BAE-watch and subsequent phylogenetic analysis, suggests that recent horizontal gene transfer between bacteria and eukaryotes has been rare. However, some unusual cases of bacterial-eukaryotic homology have been identified and are being targeted for further functional study, with the aim of using C. elegans as an infection model. • References • Tettelin H, et al. 2000. Science 287:1809-1815. • Read TD, et al. 2000. Nucleic Acids Res. 28:1397-1406. • Doolittle WF. 1998. Trends Genet. 14:307-311. • Brinkman FSL, et al. 2001. Bioinformatics. In Press. • de Koning A, et al. 2000. Mol. Biol. Evol. 17:1769-1773. • Stephens RS, et al. 1998. Science. 282:754-759. • Tan M-W et al. 1999 PNAS USA 96:715-720. • Andrew PA and Nicholas WL. 1976 Nematologica 22:451- 461. • Aballay A and Ausubel FM. 2001 PNAS USA 98:2735-2739. • Hodgkin J, et al. 2000 Curr. Biol. 10:1615-1618. • Straley SC and Perry RD. 1995 Trends Microbiol. 3:310-317. Acknowledgements This project is funded by the Peter Wall Institute for Advanced Studies