Download
1 / 1
10 likes | 110 Views
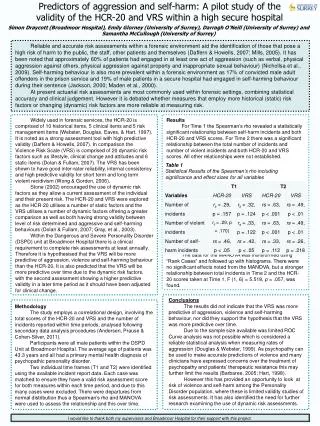
}. Four quartiles of week 8 trough concentration. Q4. Q3. Q2. Q1. Placebo.

E N D
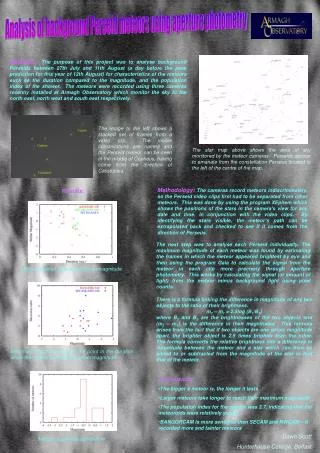
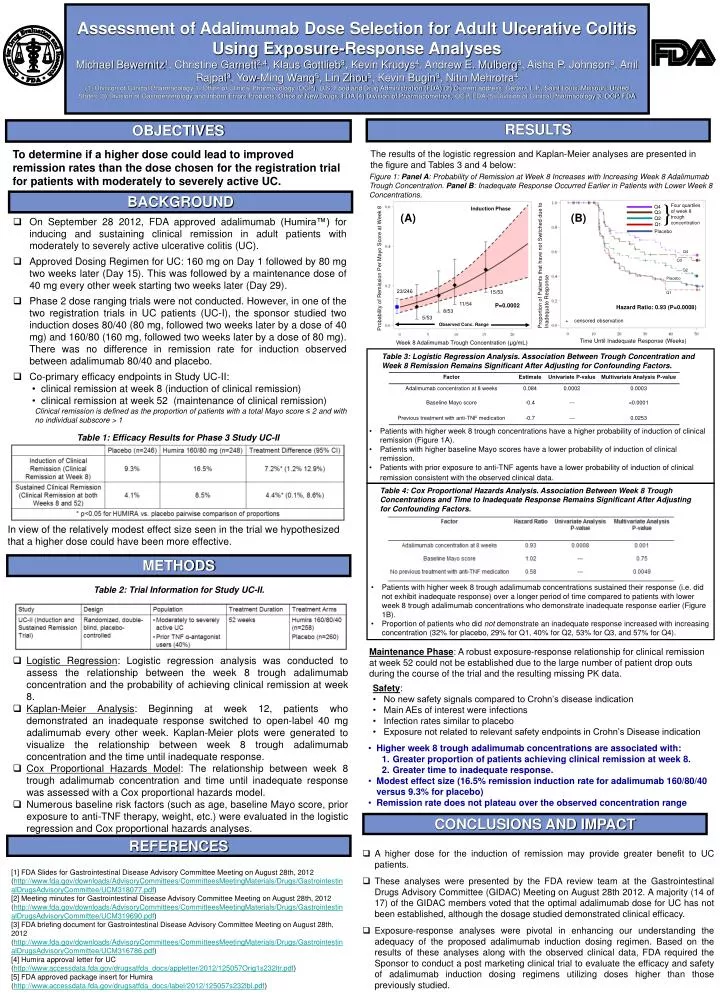
} Four quartiles of week 8 trough concentration Q4 Q3 Q2 Q1 Placebo Assessment of Adalimumab Dose Selection for Adult Ulcerative Colitis Using Exposure-Response AnalysesMichael Bewernitz1, Christine Garnett2,4, Klaus Gottlieb3, Kevin Krudys4, Andrew E. Mulberg3, Aisha P. Johnson3, Anil Rajpal3, Yow-Ming Wang5, Lin Zhou5, Kevin Bugin3, Nitin Mehrotra4(1) Division of Clinical Pharmacology 1, Office of Clinical Pharmacology (OCP), U.S. Food and Drug Administration (FDA) (2) Current address: Certara L.P., Saint Louis, Missouri, United States (3) Division of Gastroenterology and Inborn Errors Products, Office of New Drugs, FDA (4) Division of Pharmacometrics, OCP, FDA (5) Division of Clinical Pharmacology 3, OCP, FDA RESULTS OBJECTIVES To determine if a higher dose could lead to improved remission rates than the dose chosen for the registration trial for patients with moderately to severely active UC. The results of the logistic regression and Kaplan-Meier analyses are presented in the figure and Tables 3 and 4 below: Figure 1: Panel A: Probability of Remission at Week 8 Increases with Increasing Week 8 Adalimumab Trough Concentration. Panel B: Inadequate Response Occurred Earlier in Patients with Lower Week 8 Concentrations. BACKGROUND Proportion of Patients that have not Switched due to Inadequate Response Probability of Remission Per Mayo Score at Week 8 Induction Phase (A) (B) • On September 28 2012, FDA approved adalimumab (Humira™) for inducing and sustaining clinical remission in adult patients with moderately to severely active ulcerative colitis (UC). • Approved Dosing Regimen for UC: 160 mg on Day 1 followed by 80 mg two weeks later (Day 15). This was followed by a maintenance dose of 40 mg every other week starting two weeks later (Day 29). • Phase 2 dose ranging trials were not conducted. However, in one of the two registration trials in UC patients (UC-I), the sponsor studied two induction doses 80/40 (80 mg, followed two weeks later by a dose of 40 mg) and 160/80 (160 mg, followed two weeks later by a dose of 80 mg). There was no difference in remission rate for induction observed between adalimumab 80/40 and placebo. • Co-primary efficacy endpoints in Study UC-II: • clinical remission at week 8 (induction of clinical remission) • clinical remission at week 52 (maintenance of clinical remission) Clinical remission is defined as the proportion of patients with a total Mayo score ≤ 2 and with no individual subscore > 1 Q4 Q3 Q2 Placebo 23/246 15/53 Q1 11/54 P=0.0002 Hazard Ratio: 0.93 (P=0.0008) 8/53 5/53 + censored observation Observed Conc. Range Time Until Inadequate Response (Weeks) Week 8 Adalimumab Trough Concentration (μg/mL) Table 3: Logistic Regression Analysis. Association Between Trough Concentration and Week 8 Remission Remains Significant After Adjusting for Confounding Factors. • Patients with higher week 8 trough concentrations have a higher probability of induction of clinical remission (Figure 1A). • Patients with higher baseline Mayo scores have a lower probability of induction of clinical remission. • Patients with prior exposure to anti-TNF agents have a lower probability of induction of clinical remission consistent with the observed clinical data. Table 1: Efficacy Results for Phase 3 Study UC-II Table 4: Cox Proportional Hazards Analysis. Association Between Week 8 Trough Concentrations and Time to Inadequate Response Remains Significant After Adjusting for Confounding Factors. In view of the relatively modest effect size seen in the trial we hypothesized that a higher dose could have been more effective. METHODS • Patients with higher week 8 trough adalimumab concentrations sustained their response (i.e. did not exhibit inadequate response) over a longer period of time compared to patients with lower week 8 trough adalimumab concentrations who demonstrate inadequate response earlier (Figure 1B). • Proportion of patients who did not demonstrate an inadequate response increased with increasing concentration (32% for placebo, 29% for Q1, 40% for Q2, 53% for Q3, and 57% for Q4). Table 2: Trial Information for Study UC-II. Maintenance Phase: A robust exposure-response relationship for clinical remission at week 52 could not be established due to the large number of patient drop outs during the course of the trial and the resulting missing PK data. • Logistic Regression: Logistic regression analysis was conducted to assess the relationship between the week 8 trough adalimumab concentration and the probability of achieving clinical remission at week 8. • Kaplan-Meier Analysis: Beginning at week 12, patients who demonstrated an inadequate response switched to open-label 40 mg adalimumab every other week. Kaplan-Meier plots were generated to visualize the relationship between week 8 trough adalimumab concentration and the time until inadequate response. • Cox Proportional Hazards Model: The relationship between week 8 trough adalimumab concentration and time until inadequate response was assessed with a Cox proportional hazards model. • Numerous baseline risk factors (such as age, baseline Mayo score, prior exposure to anti-TNF therapy, weight, etc.) were evaluated in the logistic regression and Cox proportional hazards analyses. • Safety: • No new safety signals compared to Crohn’s disease indication • Main AEs of interest were infections • Infection rates similar to placebo • Exposure not related to relevant safety endpoints in Crohn’s Disease indication • Higher week 8 trough adalimumab concentrations are associated with: • Greater proportion of patients achieving clinical remission at week 8. • Greater time to inadequate response. • Modest effect size (16.5% remission induction rate for adalimumab 160/80/40 versus 9.3% for placebo) • Remission rate does not plateau over the observed concentration range CONCLUSIONS AND IMPACT REFERENCES • A higher dose for the induction of remission may provide greater benefit to UC patients. • These analyses were presented by the FDA review team at the Gastrointestinal Drugs Advisory Committee (GIDAC) Meeting on August 28th 2012. A majority (14 of 17) of the GIDAC members voted that the optimal adalimumab dose for UC has not been established, although the dosage studied demonstrated clinical efficacy. • Exposure-response analyses were pivotal in enhancing our understanding the adequacy of the proposed adalimumab induction dosing regimen. Based on the results of these analyses along with the observed clinical data, FDA required the Sponsor to conduct a post marketing clinical trial to evaluate the efficacy and safety of adalimumab induction dosing regimens utilizing doses higher than those previously studied. [1] FDA Slides for Gastrointestinal Disease Advisory Committee Meeting on August 28th, 2012 (http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/GastrointestinalDrugsAdvisoryCommittee/UCM318077.pdf) [2] Meeting minutes for Gastrointestinal Disease Advisory Committee Meeting on August 28th, 2012 (http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/GastrointestinalDrugsAdvisoryCommittee/UCM319690.pdf) [3] FDA briefing document for Gastrointestinal Disease Advisory Committee Meeting on August 28th, 2012 (http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/GastrointestinalDrugsAdvisoryCommittee/UCM316786.pdf) [4] Humira approval letter for UC (http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2012/125057Orig1s232ltr.pdf) [5] FDA approved package insert for Humira (http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125057s232lbl.pdf)