Download

1 / 21
210 likes | 338 Views
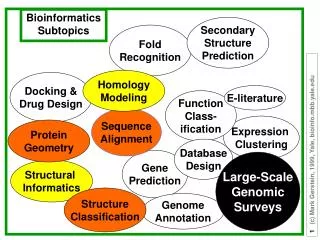
A Four-Body Statistical Potential For Protein Fold Recognition. Bala Krishnamoorthy and Alex Tropsha UNC Chapel Hill. Nov 17, 2003. Four-Body Potentials. Outline. Motivation. Hypothesis. Four-body statistical potentials. Application to folding simulations.

E N D
A Four-Body Statistical Potential For Protein Fold Recognition Bala Krishnamoorthy and Alex Tropsha UNC Chapel Hill Nov 17, 2003
Four-Body Potentials Outline Motivation Hypothesis Four-body statistical potentials Application to folding simulations Application to predictions from CASP5 and Livebench 6
Four-Body Potentials Motivation Knowledge of protein structure is essential to understand their function(s) Number of proteins (sequences known) is growing exponentially Traditional methods for determining protein structure (X-ray crystallography, NMR etc.) do not yield quick results Need to develop statistical methods that help with protein fold recognition
Four-Body Potentials Hypothesis Specific nearest neighbor residue contacts in protein structures have non-random propensities for occurrence. The propensities of occurrence of nearest neighbor clusters can be used to score compatibility between protein sequence and structure
Four-Body Potentials SNAPP Simplicial Neighborhood Analysis of Protein Packing 3-D Packing 2-D Packing 2-D:3 neighbors in mutual contact 3-D: 4 neighbor clusters
Four-Body Potentials Objective definition of the nearest neighborhood of each residue is needed Use the Voronoi diagram of the protein • gives convex hulls around each residue (represented as a point) that define the nearest neighborhood of the residue Delaunay triangulation – defined as the dual of the Voronoi diagram
Four-Body Potentials Tessellation of protein structure (in 3D) Residues are represented by their side-chain centers (or by their C-α atoms) Protein structure represented as an aggregate of space filling, non-intersecting and irregular tetrahedra Nearest neighbor residues are identified as unique sets of four residues each (tetrahedral quadruplets)
Four-Body Potentials Four-body Statistical Potentials Denote each quadruplet by { i , j , k , l } i,j,k and l can be any of the 20 amino acids Total number of possible quadruplets is 8855 AALV VALI TLKM YYYY …
Four-Body Potentials Based on the back-bone connectivity of {i,j,k,l}, there can be five types of tetrahedra (indexed as 0,1,2,3 and 4 respectively ) The propensities of the {i,j,k,l} quadruplets of each type t could be used to develop four-body statistical potentials
Four-Body Potentials f q ijkl_t = log p ijkl_t ijkl_t a p p = C a a a t i j k l ijkl_t Four-body compositional propensities of Delaunay simplices f - observed frequency of occurrence in the training set of quad {ijkl} in a type t tetrahedron ijkl_t p • expected frequency of occurrence in the training set of • residues i,j,k and l in a type t tetrahedron ijkl_t a – individual AA frequency i p – frequency of type t tetrahedra t C – combinatorial factor
Four-Body Potentials diverse training set of 1166 protein chains with known structure For a test conformation, the total log-likelihood score is calculated by adding the score for each tetrahedron in its Delaunay tessellation. Higher Score ↔ better structure
Four-Body Potentials MD Simulation of proteins Comparison of pre- and post-TS (transition) structure of CI2 vs. native CI2 * Pre-TS (six structures) Post-TS (20 structures) Native Go potentials (native structure specific) fail to discriminate between the three! *structures courtesy of Dr. E. Shaknovich, Harvard (Ref: J. Mol. Biol. 296 (2000) p1183-1188)
Four-Body Potentials Comparison of total scores for pre- and post-TS structures of CI2 vs. native CI2 N.B. - The 5th pre-TS instance actually had a 0.10 probability of folding (the other five pre-TS structures had ~ 0 probability of folding)
Four-Body Potentials Four-Body Potentials L49 I20 V47 I57 A16 V51 L8 V13 I29 V31 V13 V31 V51 L49 Structure profiles of pre-TS vs. post-TS structure of CI2 Profile ProCAM of Post-TS structure
Four-Body Potentials SNAPP analysis of pre-TS vs. post-TS structure of CI2 Pre-TS Post-TS
Four-Body Potentials I48 A37 F18 L16 Y8 W35 Y52 G46 Structure profiles of pre-TS vs. post-TS structure of SH3
Four-Body Potentials Scoring Livebench 6 and CASP5 predictions Livebench Automated evaluation of structure prediction servers Set 6 had 32 “easy” and 66 “hard” targets CASP 5 3D coordinate models submitted for 56 targets Native structure of 33 targets has been released - rank 3D predictions using four-body potentials - compare with the ranking using global structural similarity measures (like MaxSub)
Four-Body Potentials To compare rankings, use predictive index (PI) Here, E – experimental values, P – predicted values
Four-Body Potentials Livebench 6 10 models for each target made by PMODELLER PI for 28 “easy” targets and 38 “hard” targets (at least one model had a non-zero MaxSub score)
Four-Body Potentials CASP 5 For 18 targets (out of 33), the native structure ranked better than allpredictions For 26 (out of 33) targets, the native structure was ranked within the top 3.5 % of all the predictions
Four-Body Potentials Conclusions A four-body statistical scoring function is developed based on the Delaunay tessellation of proteins Discriminates native from decoy structures in most of the cases Distinguishes pre- and post-transition state structures and the native structure from MD folding simulation trajectories Highly effective in the accurate ranking of Livebench 6 and CASP5 predictions