Download

1 / 22
230 likes | 254 Views
Explore protein structures & functions using machine learning techniques for better insights. Learn about quaternary folds & complex protein networks.

E N D
Protein Tertiary and Quaternary Fold Recognition: A ML Approach Jaime Carbonell Joint work with: Yan Liu(IBM), Vanathi Gopalakrishnan (U Pitt), Peter Weigele (MIT) Language Technologies Institute Carnegie Mellon University Machine Learning Lunch – 11-April-2007
Nobelprize.org DSCTFTTAAAAKAGKAKAG Protein sequence + Protein function Protein structure Snapshot of Cell Biology
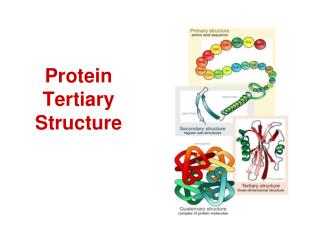
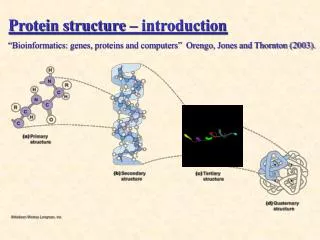
(Borrowed from: Judith Klein-Seetharaman) PROTEINS Sequence Structure Function Primary Sequence MNGTEGPNFY VPFSNKTGVV RSPFEAPQYY LAEPWQFSML AAYMFLLIML GFPINFLTLY VTVQHKKLRT PLNYILLNLA VADLFMVFGG FTTTLYTSLH GYFVFGPTGC NLEGFFATLG GEIALWSLVV LAIERYVVVC KPMSNFRFGE NHAIMGVAFT WVMALACAAP PLVGWSRYIP EGMQCSCGID YYTPHEETNN ESFVIYMFVV HFIIPLIVIF FCYGQLVFTV KEAAAQQQES ATTQKAEKEV TRMVIIMVIA FLICWLPYAG VAFYIFTHQG SDFGPIFMTI PAFFAKTSAV YNPVIYIMMN KQFRNCMVTT LCCGKNPLGD DEASTTVSKT ETSQVAPA Folding 3D Structure Complex function within network of proteins Normal
Disease PROTEINS Sequence Structure Function Primary Sequence MNGTEGPNFY VPFSNKTGVV RSPFEAPQYY LAEPWQFSML AAYMFLLIML GFPINFLTLY VTVQHKKLRT PLNYILLNLA VADLFMVFGG FTTTLYTSLH GYFVFGPTGC NLEGFFATLG GEIALWSLVV LAIERYVVVC KPMSNFRFGE NHAIMGVAFT WVMALACAAP PLVGWSRYIP EGMQCSCGID YYTPHEETNN ESFVIYMFVV HFIIPLIVIF FCYGQLVFTV KEAAAQQQES ATTQKAEKEV TRMVIIMVIA FLICWLPYAG VAFYIFTHQG SDFGPIFMTI PAFFAKTSAV YNPVIYIMMN KQFRNCMVTT LCCGKNPLGD DEASTTVSKT ETSQVAPA Folding 3D Structure Complex function within network of proteins
Example Protein Structures Triple beta-spiral fold in Adenovirus Fiber Shaft Adenovirus Fibre Shaft Virus Capsid
Predicting Protein Structures • Protein Structure is a key determinant of protein function • Crystalography to resolve protein structures experimentally in-vitro is very expensive, NMR can only resolve very-small proteins • The gap between the known protein sequences and structures: • 3,023,461 sequences v.s. 36,247 resolved structures (1.2%) • Therefore we need to predict structures in-silico
Seq 1: APA FSVSPA … SGACGPECAESG Seq 2 : DSCTFT…TAAAAKAGKAKCSTITL Quaternary Folds and Alignments • Protein fold • Identifiable regular arrangement of secondary structural elements • Thus far, a limited number of protein folds have been discovered (~1000) • Very few research work on quaternary folds • Complex structures and few labeled data • Quaternary fold recognition
Previous Work • Sequence similarity perspective • Sequence similarity searches, e.g. PSI-BLAST [Altschul et al, 1997] • Profile HMM, .e.g. HMMER [Durbin et al, 1998] and SAM [Karplus et al, 1998] • Window-based methods, e.g. PSI_pred [Jones, 2001] • Physical forces perspective • Homology modeling or threading, e.g. Threader [Jones, 1998] • Structural biology perspective • Painstakingly hand-engineered methods for specific structures, e.g.αα- and ββ- hairpins, β-turn and β-helix [Efimov, 1991; Wilmot and Thornton, 1990; Bradley at al, 2001] Fail to capture the structure properties and long-range dependencies Generative models based on rough approximation of free-energy, perform very poorly on complex structures Very Hard to generalize due to built-in constants, fixed features
Conditional Random Fields • Hidden Markov model (HMM)[Rabiner, 1989] • Conditional random fields (CRFs)[Lafferty et al, 2001] • Model conditional probability directly (discriminative models, directly optimizable) • Allow arbitrary dependencies in observation • Adaptive to different loss functions and regularizers • Promising results in multiple applications • But, need to scale up (computationally) and extend to long-distance dependencies
Our Solution: Conditional Graphical Models Local dependency Long-range dependency • Outputs Y = {M, {Wi} }, where Wi = {pi, qi, si} • Feature definition • Node feature • Local interaction feature • Long-range interaction feature
Joint Labels Linked Segmentation CRF • Node: secondary structure elements and/or simple fold • Edges: Local interactions and long-range inter-chain and intra-chain interactions • L-SCRF: conditional probability of y given x is defined as
Linked Segmentation CRF (II) • Classification: • Training : learn the model parametersλ • Minimizing regularized negative log loss • Iterative search algorithms by seeking the direction whose empirical values agree with the expectation • Complex graphs results in huge computational complexity
Approximate Inference of L-SCRF • Most approximation algorithms cannot handle variable number of nodes in the graph, but we need variable graph topologies, so… • Reversible jump MCMC sampling [Greens, 1995, Schmidler et al, 2001] withFour types of Metropolis operators • State switching • Position switching • Segment split • Segment merge • Simulated annealing reversible jump MCMC [Andireu et al, 2000] • Replace the sample with RJ MCMC • Theoretically converge on the global optimum
Tertiary Fold Recognition: β-Helix fold • Histogram and ranks for known β-helices against PDB-minus dataset 5 Chain graph model reduces the real running time of SCRFs model by around 50 times
Fold Alignment Prediction:β-Helix • Predicted alignment for known β-helices on cross-family validation
Discovery of New Potential β-helices • Run structural predictor seeking potential β-helices from Uniprot (structurally unresolved) databases • Full list (98 new predictions) can be accessed at www.cs.cmu.edu/~yanliu/SCRF.html • Verification on 3 proteins with later experimentally resolved structures from different organisms • 1YP2: Potato Tuber ADP-Glucose Pyrophosphorylase • 1PXZ: The Major Allergen From Cedar Pollen • GP14 of Shigella bacteriophage as a β-helix protein • No single false positive!
Experiments: Target Quaternary Fold • Triple beta-spirals [van Raaij et al. Nature 1999] • Virus fibers in adenovirus, reovirus and PRD1 • Double barrel trimer [Benson et al, 2004] • Coat protein of adenovirus, PRD1, STIV, PBCV
Experiment Results: Fold Recognition Triple beta-spirals Double barrel-trimer
Triple beta-spirals Four states: B1, B2, T1 and T2 Correct Alignment: B1: i – o B2: a - h Predicted Alignment B1 B2 Experiment Results: Alignment Prediction
Experiment Results:Discovery of New Membership Proteins • Predicted membership proteins of triple beta-spirals can be accessed at http://www.cs.cmu.edu/~yanliu/swissprot_list.xls • Membership proteins of double barrel-trimer suggested by biologists [Benson, 2005] compared with L-SCRF predictions
Concluding Remarks • Conditional graphical models for protein structure prediction • Effective representation for protein structural properties • Feasibility to incorporate different kinds of informative features • Efficient inference algorithms for large-scale applications • A major extension compared with previous work • Knowledge representation through graphical models • Ability to handle long-range interactions within one chain and between chains • Future work • Automatic learning of graph topology • Active learning – including minority-class discovery